
Patiromer
1260643-52-4 FREE FORM
CAS 1208912-84-8
(C10 H10 . C8 H14 . C3 H3 F O2 . 1/2 Ca)x
2-Propenoic acid, 2-fluoro-, calcium salt (2:1), polymer with diethenylbenzene and 1,7-octadiene
RLY5016
RELYPSA INNOVATOR
Veltassa is a powder for suspension in water for oral administration. The active ingredient is patiromer sorbitex calcium which consists of the active moiety, patiromer, a non-absorbed potassium-binding polymer, and a calcium-sorbitol counterion.

Each gram of patiromer is equivalent to a nominal amount of 2 grams of patiromer sorbitex calcium. The chemical name for patiromer sorbitex calcium is cross-linked polymer of calcium 2-fluoroprop-2-enoate with diethenylbenzene and octa-1,7-diene, combination with D-glucitol.
Mechanism of Action
Veltassa is a non-absorbed, cation exchange polymer that contains a calcium-sorbitol counterion. Veltassa increases fecal potassium excretion through binding of potassium in the lumen of the gastrointestinal tract. Binding of potassium reduces the concentration of free potassium in the gastrointestinal lumen, resulting in a reduction of serum potassium levels.
Treatment of Hyperkalemia
Hyperkalemia is usually asymptomatic but occasionally can lead to life-threatening cardiac arrhythmias and increased all-cause and in-hospital mortality, particularly in patients with CKD and associated cardiovascular diseases (Jain et al., 2012; McMahon et al., 2012; Khanagavi et al., 2014). However, there is limited evidence from randomized clinical trials regarding the most effective therapy for acute management of hyperkalemia (Khanagavi et al., 2014) and a Cochrane analysis of emergency interventions for hyperkalemia found that none of the studies reported mortality or cardiac arrhythmias, but reports focused on PK (Mahoney et al., 2005). Thus, recommendations are based on opinions and vary with institutional practice guidelines (Elliot et al., 2010; Khanagavi et al., 2014). Management of hyperkalemia includes reducing potassium intake, discontinuing potassium supplements, treatment of precipitating risk factors, and careful review of prescribed drugs affecting potassium homeostasis. Treatment of life-threatening hyperkalemia includes nebulized or inhaled beta-agonists (albuterol, salbutamol) or intravenous (IV) insulin-and-glucose, which stimulate intracellular potassium uptake, their combination being more effective than either alone. When arrhythmias are present, IV calcium might stabilize the cardiac resting membrane potential. Sodium bicarbonate may be indicated in patients with severe metabolic acidosis. Potassium can be effectively eliminated by hemodialysis or increasing its renal (loop diuretics) and gastrointestinal (GI) excretion with sodium polystyrene sulfonate, an ion-exchange resin that exchanges sodium for potassium in the colon. However, this resin produces serious GI adverse events (ischemic colitis, bleeding, perforation, or necrosis). Therefore, there is an unmet need of safer and more effective drugs producing a rapid and sustained PK reduction in patients with hyperkalemia.
In this article we review two new polymer-based, non-systemic oral agents, patiromer calcium (RLY5016) and zirconium silicate (ZS-9), under clinical development designed to induce potassium loss via the GI tract, particularly the colon, and reduce PK in patients with hyperkalemia.
1. Patiromer calcium
This metal-free cross-linked fluoroacrylate polymer (structure not available) exchanges cations through the gastrointestinal (GI) tract. It preferentially binds soluble potassium in the colon, increases its fecal excretion and reduces PK under hyperkalemic conditions.
The development program of patiromer includes several clinical trials. An open-label, single-arm study evaluated a titration regimen for patiromer in 60 HF patients with CKD treated with ACEIs, ARBs, or beta blockers (clinicaltrials.gov identifier: NCT01130597). Another open-label, randomized, dose ranging trial determined the optimal starting dose and safety of patiromer in 300 hypertensive patients with diabetic nephropathy treated with ACEIs and/or ARBs, with or without spironolactone (NCT01371747). The primary outcomes were the change in PK from baseline to the end of the study. Unfortunately, the results of these trials were not published.
In a double-blind, placebo-controlled trial (PEARL-HF, NCT00868439), 105 patients with a baseline PK of 4.7 mmol/L and HF (NYHA class II-III) treated with spironolactone in addition to standard therapy were randomized to patiromer (15 g) or placebo BID for 4 weeks (Pitt et al., 2011). Spironolactone, initiated at 25 mg/day, was increased to 50 mg/day on day 15 if PK was ≤5.1 mmol/L. Patients were eligible for the trial if they had either CKD (eGFR <60 ml/min) or a history of hyperkalemia leading to discontinuation of RAASIs or beta-blockers. Compared with placebo, patiromer decreased the PK (-0.22 mmol/L, while PK increased in the placebo group +0.23 mmol/L, P<0.001), and the incidence of hyperkalemia (7% vs. 25%, P=0.015) and increased the number of patients up-titrated to spironolactone 50 mg/day (91% vs. 74%, P=0.019). A similar reduction in PK and hyperkalemia was observed in patients with an eGFR <60 ml/min. Patiromer produced more GI adverse events (flatulence, diarrhea, constipation, vomiting: 21% vs 6%), hypokalemia (<4.0 mmol/L: 47% vs 10%, P<0.001) and hypomagnesaemia (<1.8 mg/dL: 24% vs. 2.1%), but similar adverse events leading to study discontinuation compared to placebo. Unfortunately, recruited patients had normokalemia and basal eGFR in the treatment group was 84 ml/min. Thus, this study did not answer whether patiromer is effective in reducing PK in patients with CKD and/or HF who develop hyperkalemia on RAASIs.
A two-part phase 3 study evaluated the efficacy and safety of patiromer in the treatment of hyperkalemia (NCT01810939). In a single-blind phase (part A) 243 patients with hyperkalemia and CKD (102 with HF) on RAASIs were treated with patiromer BID for 4 weeks: 4.2 g in patients with mild hyperkalemia (5.1-<5.5 mmol/L, n=92) and 8.4 g in patients with moderate-to-severe hyperkalemia (5.5-<6.5 mmol/L, n=151). Part B was a placebo-controlled, randomized, withdrawal phase designed to confirm the maintained efficacy of patiromer and the recurrent hyperkalemia following that drug’s withdrawal. Patients (n=107) who completed phase A with a normal PK were randomized to continue on patiromer (27 with HF) or placebo (22 with HF) besides RAASIs for 8 weeks. The primary endpoint was the difference in mean PK between the patiromer and placebo groups from baseline to the end of the study or when the patient first had a PK <3.8 or ≥5.5 mmol/L. In part A patiromer produced a rapid reduction in PK that persisted throughout the study in patients with and without HF (-1.06 and -0.98 mmol/L, respectively; both P<0.001 vs. placebo); three-fourths of patients in both groups had normal PK (3.8-<5.1 mmol/L) at 4 weeks. In part B patiromer reduced PK (-0.64 mmol/L) in patients with or without HF (P<0.001). As compared with placebo, fewer patients, with or without HF, presented recurrent hyperkalemia in the patiromer group or required RAASI discontinuation regardless of HF status (Pitt, 2014). Patiromer was well-tolerated, with a safety profile similar to placebo even in HF patients. The most common adverse events were nausea, diarrhea, and hypokalemia.
INDICATIONS AND USAGE
Veltassa is a potassium binder indicated for the treatment of hyperkalemia.Veltassa should not be used as an emergency treatment for lifethreatening hyperkalemia because of its delayed onset of action.
Patiromer (USAN, trade name Veltassa) is a drug used for the treatment of hyperkalemia (elevated blood potassium levels), a condition that may lead to palpitations and arrhythmia (irregular heartbeat). It works by binding potassium in the gut.[1][2]

Medical uses
Patiromer is used for the treatment of hyperkalemia, but not as an emergency treatment for life-threatening hyperkalemia, because it acts relatively slowly.[2] Such a condition needs other kinds of treatment, for example calcium infusions, insulin plus glucose infusions, salbutamol inhalation, and hemodialysis.[3]Typical reasons for hyperkalemia are renal insufficiency and application of drugs that inhibit the renin–angiotensin–aldosterone system (RAAS) – e.g. ACE inhibitors, angiotensin II receptor antagonists, or potassium-sparing diuretics – or that interfere with renal function in general, such as nonsteroidal anti-inflammatory drugs (NSAIDs).[4][5]
Adverse effects
Patiromer was generally well tolerated in studies. Side effects that occurred in more than 2% of patients included in clinical trials were mainly gastro-intestinal problems such as constipation, diarrhea, nausea, and flatulence, and also hypomagnesemia (low levels of magnesium in the blood) in 5% of patients, because patiromer binds magnesium in the gut as well.[2][6]Interactions
No interaction studies have been done in humans. Patiromer binds to many substances besides potassium, including numerous orally administered drugs (about half of those tested in vitro). This could reduce their availability and thus effectiveness,[2] wherefore patiromer has received a boxed warning by the US Food and Drug Administration (FDA), telling patients to wait for at least six hours between taking patiromer and any other oral drugs.[7]Pharmacology
Mechanism of action
Patiromer works by binding free potassium ions in the gastrointestinal tract and releasing calcium ions for exchange, thus lowering the amount of potassium available for absorption into the bloodstream and increasing the amount that is excreted via the feces. The net effect is a reduction of potassium levels in the blood serum.[2][4]Lowering of potassium levels is detectable 7 hours after administration. Levels continue to decrease for at least 48 hours if treatment is continued, and remain stable for 24 hours after administration of the last dose. After this, potassium levels start to rise again over a period of at least four days.[2]
Pharmacokinetics
Patiromer is not absorbed from the gut, is not metabolized, and is excreted in unchanged form with the feces.[2]Physical and chemical properties
The substance is a cross-linked polymer of 2-fluoroacrylic acid (91% in terms of amount of substance) with divinylbenzenes (8%) and 1,7-octadiene (1%). It is used in form of its calcium salt (ratio 2:1) and with sorbitol (one molecule per two calcium ions or four fluoroacrylic acid units), a combination called patiromer sorbitex calcium.[8]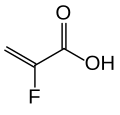 2-fluoroacrylic acid
2-fluoroacrylic acid o-divinylbenzene
o-divinylbenzene p-divinylbenzene
p-divinylbenzene 1,7-octadiene
1,7-octadiene


Hyperkalemia Is a Clinical Challenge
Hyperkalemia may result from increased potassium intake, impaired distribution between the intracellular and extracellular spaces, and/or conditions that reduce potassium excretion, including CKD, hypertension, diabetes mellitus, or chronic heart failure (HF) (Jain et al., 2012). Additionally, drugs and nutritional/herbal supplements (Table 1) can produce hyperkalemia in up to 88% of hospitalized patients by impairing normal potassium regulation (Hollander-Rodríguez and Calvert, 2006; Khanagavi et al., 2014).
 Although
the prevalence of hyperkalemia in the general population is unknown, it
is present in 1-10% of hospitalized patients depending on how
hyperkalemia is defined (McMahon et al., 2012; Gennari, 2002).
Hyperkalemia is a common problem in patients with conditions that reduce
potassium excretion, especially when treated with beta-adrenergic
blockers that inhibit Na+,K+-ATPase activity or
RAAS inhibitors (RAASIs) [angiotensin-converting-enzyme inhibitors
(ACEIs), angiotensin receptor blockers (ARBs), mineralocorticoid
receptor antagonists or renin inhibitors] that decrease aldosterone
excretion (Jain et al., 2012; Weir and Rolfe, 2010). The
incidence of hyperkalemia with RAASIs in monotherapy is low (≤2%) in
patients without predisposing factors, but increases with dual RAASIs
(5%) and in patients with risk factors such as CKD, HF, and/or diabetes
(5-10%) (Weir and Rolfe, 2010). Thus, hyperkalemia is a key limitation
to fully titrate RAASIs in these patients who are most likely to benefit
from treatment. Thus, we need new drugs to control hyperkalemia in
these patients while maintaining the use of RAASIs.
Although
the prevalence of hyperkalemia in the general population is unknown, it
is present in 1-10% of hospitalized patients depending on how
hyperkalemia is defined (McMahon et al., 2012; Gennari, 2002).
Hyperkalemia is a common problem in patients with conditions that reduce
potassium excretion, especially when treated with beta-adrenergic
blockers that inhibit Na+,K+-ATPase activity or
RAAS inhibitors (RAASIs) [angiotensin-converting-enzyme inhibitors
(ACEIs), angiotensin receptor blockers (ARBs), mineralocorticoid
receptor antagonists or renin inhibitors] that decrease aldosterone
excretion (Jain et al., 2012; Weir and Rolfe, 2010). The
incidence of hyperkalemia with RAASIs in monotherapy is low (≤2%) in
patients without predisposing factors, but increases with dual RAASIs
(5%) and in patients with risk factors such as CKD, HF, and/or diabetes
(5-10%) (Weir and Rolfe, 2010). Thus, hyperkalemia is a key limitation
to fully titrate RAASIs in these patients who are most likely to benefit
from treatment. Thus, we need new drugs to control hyperkalemia in
these patients while maintaining the use of RAASIs.History
Studies
In a Phase III multicenter clinical trial including 237 patients with hyperkalemia under RAAS inhibitor treatment, 76% of participants reached normal serum potassium levels within four weeks. After subsequent randomization of 107 responders into a group receiving continued patiromer treatment and a placebo group, re-occurrence of hyperkalemia was 15% versus 60%, respectively.[9]Approval
The US FDA approved patiromer in October 2015.[7] The drug is not approved in Europe as of January 2016.
PATENT
PATENT
References
- 1 Henneman, A; Guirguis, E; Grace, Y; Patel, D; Shah, B (2016). "Emerging therapies for the management of chronic hyperkalemia in the ambulatory care setting". American Journal of Health-System Pharmacy 73 (2): 33–44. doi:10.2146/ajhp150457. PMID 26721532.
- 2FDA Professional Drug Information for Veltassa.
- 3Vanden Hoek TL, Morrison LJ, Shuster M, Donnino M, Sinz E, Lavonas EJ, Jeejeebhoy FM, Gabrielli A; Morrison; Shuster; Donnino; Sinz; Lavonas; Jeejeebhoy; Gabrielli (2010-11-02). "Part 12: cardiac arrest in special situations: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care". Circulation 122 (18 Suppl 3): S829–61. doi:10.1161/CIRCULATIONAHA.110.971069. PMID 20956228.
- 4Esteras, R.; Perez-Gomez, M. V.; Rodriguez-Osorio, L.; Ortiz, A.; Fernandez-Fernandez, B. (2015). "Combination use of medicines from two classes of renin-angiotensin system blocking agents: Risk of hyperkalemia, hypotension, and impaired renal function". Therapeutic Advances in Drug Safety 6 (4): 166. doi:10.1177/2042098615589905. PMID 26301070.
- 5Rastegar, A; Soleimani, M (2001). "Hypokalaemia and hyperkalaemia". Postgraduate Medical Journal 77 (914): 759–64. doi:10.1136/pmj.77.914.759. PMC 1742191. PMID 11723313.
- 6Tamargo, J; Caballero, R; Delpón, E (2014). "New drugs for the treatment of hyperkalemia in patients treated with renin-angiotensin-aldosterone system inhibitors -- hype or hope?". Discovery medicine 18 (100): 249–54. PMID 25425465.
- 7"FDA approves new drug to treat hyperkalemia". FDA. 21 October 2015.
- 8RxList: Veltassa.
- 9Weir, Matthew R.; Bakris, George L.; Bushinsky, David A.; Mayo, Martha R.; Garza, Dahlia; Stasiv, Yuri; Wittes, Janet; Christ-Schmidt, Heidi; Berman, Lance; Pitt, Bertram (2015). "Patiromer in Patients with Kidney Disease and Hyperkalemia Receiving RAAS Inhibitors". New England Journal of Medicine 372 (3): 211. doi:10.1056/NEJMoa1410853. PMID 25415805.
 | |
| Systematic (IUPAC) name | |
|---|---|
2-Fluoropropenoic acid, cross-linked polymer with diethenylbenzene and 1,7-octadiene
| |
| Clinical data | |
| Trade names | Veltassa |
| AHFS/Drugs.com | entry |
| Legal status |
|
| Routes of administration | Oral suspension |
| Pharmacokinetic data | |
| Bioavailability | Not absorbed |
| Metabolism | None |
| Onset of action | 7 hrs |
| Duration of action | 24 hrs |
| Excretion | Feces |
| Identifiers | |
| CAS Number | 1260643-52-4 1208912-84-8 (calcium salt) |
| ATC code | None |
| PubChem | SID 135626866 |
| DrugBank | DB09263 |
| UNII | 1FQ2RY5YHH |
| KEGG | D10148 |
| ChEMBL | CHEMBL2107875 |
| Synonyms | RLY5016 |
| Chemical data | |
| Formula | [(C3H3FO2)182·(C10H10)8·(C8H14)10]n [Ca91(C3H2FO2)182·(C10H10)8·(C8H14)10]n (calcium salt) |














{kind=link}