Synthesis of Analogs of Arylpyrrole Antimalarial Drug Leads
Abstract
Malaria continues to be one of the most widespread infectious diseases and with recent focus on global eradication and the continual evolution of drug resistant parasitic strains, the search for potent new antimalarials has gained momentum. In contrast to rational drug design approaches, the high-throughput screening of large compound libraries for potency against the parasite in whole cell assays has identified the Arylpyrrole series as a promising chemical lead. Before looking to comprehend the complex modes of biological action, the evaluation of analogs within this new drug class and the validation of their organic synthesis may act to identify specific compounds with increased potency or chemical moieties that lend themselves to the optimisation of the drug synthesis process. The collaborative efforts of open source drug discovery may help to identify potent compounds with a high-yielding and cost effective manufacturing process and thus accelerate the development of a promising candidate for integration into clinical practice.
Introduction
Malaria is a widespread tropical infectious disease caused by a number of species of the protozoan parasite of the genus plasmodium, most notably P. falciparum, which are transmitted through the saliva of the anopheles mosquito. Per year, more than 240 million people suffer from the disease and approximately 40% of the global population live in countries where Malaria is endemic.[1] As both a consequence of and a great contributor towards poverty, Malaria costs sub-Saharan Africa alone US$12 billion in productivity every year and while an approximate cost limitation for one antimalarial treatment is US $1[1], current estimates suggest that many may only be able to afford 10 cents per treatment.[2] Thus with the majority of sufferers of a poor socioeconomic background, drug design should focus on the cost-effective synthesis of highly potent compounds, in order to minimise the quantities administered.
Driven by a knowledge of the biology of the parasite, conventionally rational drug design deduces leads based on first identifying a possible molecular target and the potential mechanisms of drug action at that target. However through reversing this process, a high-throughput screening for potent compounds in whole cell assays can act to expedite the lead identification process.[3] By this approach a single compound may be applicable to many ambiguous or poorly understood, more complex biological targets, decreasing the chances of a parasitic strain evolving resistance mechanisms at every target. Ultimately this approach will allow the rapid identification of leads which, because of their capacity to act on complex targets, may result in more effective prophylactic or therapeutic drugs which retain their activity for longer.
Recently the data from the high-throughput screening of a two million compound library was released by GlaxoSmithKline (GSK), identifying 13, 533 compounds, which inhibited the intra-erythrocytic cycle of multidrug resistant strains of P. falciparum by more than 70%. This identified several promising classes of compounds which retained low cytotoxicity to human cell lines.[4] [5] Among these, the arylpyrrole series has been isolated and the resynthesis and validation of a number of lead compounds within this series are being conducted using open source science. Disseminating information over many online mediums has encouraged collaborative efforts within the international scientific community and this free exchange of data and ideas between organisations, individuals and academic institutions may stimulate faster optimisation and development of leads.
Initial validation of arylpyrrole leads involved the resynthesis of two known series compounds, that is TCMDC 123812 and TCMDC 123794, as high quality starting points for lead optimisation with reasonable druggability.[6] Following evaluation of two possible synthesis pathways to these final compounds, slight changes were introduced to the chemical substructures and synthesis re-evaluated. Based on knowledge of the in vivo functionality of certain moieties, analogs were identified that may influence synthesis or beneficially contribute to the overall drug profile and efficacy.
Modifications to the para-position of aryl ring were evaluated, with hydrogen, methyl and trifluoromethyl groups being chosen to substitute the fluorine present in the original compounds. The introduction of bonds weaker than the original carbon-fluorine bond at that position may influence metabolic stability, influencing the affinity of the drug to para-hydroxylation.[7] However, and especially in the instance of a hydrogen in that position, a null result may be invaluable in enabling the prioritisation of other more stable functional groups. Thus antiplasmodial activity and cytotoxicity may be impacted upon as variations may influence the modes of action, changing the in vivo efficacy of the drug. Resultant changes in the physical or chemical properties of analogs may affect the potential for developing high-yielding, efficient and cost effective synthesis strategies. However ramifications of these chemical modifications on the ease of organic synthesis are hard to predict and thus the viability of the synthesis of a range of analogs must be evaluated as primary steps in the lead optimisation process.
Two potential synthesis pathways were identified for obtaining the carboxylic acid which then undergoes the addition of an amide group through a coupling reaction to achieve the final compound.[6] The first relied on a Paal Knorr cyclisation reaction between a dicarbonyl compound and a para-substituted aniline resulting in the arylpyrrole core. A Vilsmeier-Haack reaction then employed the use of phosphoryl chloride and dimethylformamide to form a Vilsmeier reagent or chloroimminium ion, which when substituted onto the pyrrole ring acts as a formylating agent to produce an aldehyde, once hydrolysed at a low pH. A second synthesis pathway involved synthesis of an ester, initially involving alkylation using a reactive enolate ion to form an intermediate before continuing with a condensation reaction into the characteristic arylpyrrole core. This report will describe the synthesis of each analog via these two alternative synthesis pathways. (Fig. 1)

Materials and Methods
1. Paal Knorr Synthesis of 2,5-dimethyl-1H-phenyl-pyrrole

2. Paal Knorr synthesis of 2,5-dimethyl-1H-(p-tolyl)- pyrrole

3. Paal Knorr Synthesis of 2,5-dimethyl -1H-(p-trifluoromethyl)phenyl-pyrrole

4. Vilsmeier-Haack synthesis of 2,5-dimethyl-1H-phenyl pyrrole-3-carboxaldehyde

5. Vilsmeier-Haack Synthesis of 2,5-dimethyl-1H-(p-tolyl)-pyrrole-3-carboxaldehyde.

6. Synthesis of ethyl 2,5-dimethyl-1-(p-tolyl)-1H-pyrrole-3-carboxylate

7. Synthesis of ethyl 2,5-dimethyl-1-phenyl-1H-pyrrole-3-carboxylate

8. Synthesis of ethyl 2,5-dimethyl-1-[p-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate
![Figure 9: Synthesis of ethyl 2,5-dimethyl-1-[p-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate](http://openwetware.org/wiki/Image:LMW_9-1.png)
General Reaction Mechanisms


Results
1. Paal-Knorr Synthesis of 2,5-dimethyl-1H-phenyl-pyrrole
Yield: 6.2 g (60.4%)
Comment: Product crystallised easily. Reasonably pure red brown granular substance after recrystallization.
2. Paal-Knorr synthesis of 2,5-dimethyl-1H-(p-tolyl)- pyrrole
Yield: 8.3 g (74.5%)
Comment: Crystallisation required use of activated charcoal. Recrystallisation produced ochre-coloured sand-like, relatively pure compound at a reasonable yield.
3. Paal-Knorr Synthesis of 2,5-dimethyl -1H-(p-trifluoromethyl)phenyl-pyrrole
Yield: 6.08g (60%)
Comment: Reaction took slightly longer. Recrystallisation produced a relatively pure red-brown sand-like compound.
4. Vilsmeier-Haack synthesis of 2,5-dimethyl-1H-phenyl pyrrole-3-carboxaldehyde
Yield: 407 mg (64%)
Comment: Vilsmeier reaction completed reasonably quickly. Pure compound that crystallised easily produced after recrystallization. (grey/beige free-flowing powder)
5. Vilsmeier-Haack Synthesis of 2,5-dimethyl-1H-(p-tolyl)-pyrrole-3-carboxaldehyde.
Yield: 423 mg (71%)
Comment: Recrystallised fairly easily producing a fairly pure grey/brown lumpy powder.
6. Synthesis of ethyl 2,5-dimethyl-1-(p-tolyl)-1H-pyrrole-3-carboxylate
Yield: 1.8 g (45.6%)
Comment: Upon the use of both activated charcoal and column chromatography, product crystallised easily to a relatively pure bright yellow powder.
7. Synthesis of ethyl 2,5-dimethyl-1-phenyl-1H-pyrrole-3-carboxylate
Yield: 952 mg (approximate yield 37%)
Comment: Lack of separation between product and impurity in column chromatography resulted in product not crystallising.
8. Synthesis of ethyl 2,5-dimethyl-1-[p-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate
Yield: 1.85 g (55%)
Comment: Crystallised easily following column chromatography to form a pale yellow powder.
Discussion
Synthesis via each reaction pathway was viable for most analogs however procedures need to be optimised and due to the nature of the corresponding reactions to the carboxylic acid, synthesis via the ester could be considered the most favourable.
The synthesis of the aldehyde via a Vilsmeier Haack synthesis following a Paal Knorr cyclisation was viable and the products synthesised achieved moderate yields and a reasonable level of purity with minimal purification steps. While viable, procedures could still be optimised further, however the oxidation of the aldehyde to obtain the carboxylic acid has proved problematic for the original resynthesis using the p-fluoro-phenyl-pyrrole-carbaldehyde. Unless a forgiving oxidation reaction becomes apparent, the simple base-hydrolysis via saponification necessary for the ester could prove a more favourable approach. For this reason only two aldehydes were synthesised with this strategy.
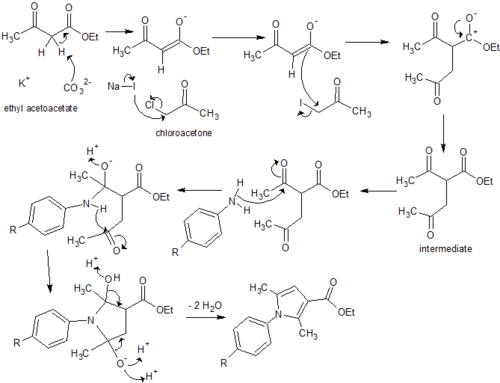
Although the ester exists in the correct oxidation state which acts to mitigate further issues with the synthesis of the carboxylic acid, complications in purification steps need to be overcome before the synthesis of analogs could be considered practical using this pathway. Potassium carbonate acts to remove a labile hydrogen off the alpha carbon of ethyl acetoacetate, which forms a reactive enolate ion. Sodium iodide substitutes the chlorine of the chloroacetone, which becomes susceptible to nucleophilic attack and an alkylation reaction follows to form a reactive intermediate. Enolate ion formation is susceptible to side reactions such as alkylation at the oxygen and while alkylation of the alpha carbon is preferential, the synthesis of the intermediate may need to be refined to ensure minimal side products and impurities. Additionally immediate condensation of the crude intermediate with the aniline derivatives may minimise formation of further side products. All three analogs required column chromatography, resulting in a significant amount of product loss and diminishing from the simplicity and practicality required for large-scale drug manufacture. It must be noted, however, this condensation reaction proved forgiving in the resynthesis of the original compounds, achieving a reasonable yield with minimal purification steps.[8] Thus appropriate optimisation of the synthesis procedures could affirm this as the most practical pathway for each analog.
Given synthesis complications and a likely propensity for para-hydroxylation, it may be unlikely that the N-phenyl analogs present promising drug leads, however further synthesis trials and testing against the parasite are required to conclusively determine this. Aqueous extraction of the final ester was problematic as a result of increased miscibility with dilute citric acid, and unsuccessful separation of product and impurity in column chromatography prevented crystallisation. It is unclear at this stage whether the parasubstituted methyl or trifluoromethyl groups retain any steric, inductive or other effects that contribute to any physicochemical properties that lend themselves to improving organic synthesis or in vivo drug targeting. The synthesis strategies require optimisation to eliminate time consuming and costly purification steps and the proceeding hydrolysis and coupling reactions need validation before testing the in vivo efficacy of the drugs in whole cell assays. Alternative reaction conditions, reagents, solvents or even new synthesis pathways should be tested to achieve optimisation as, irrespective of the drug’s antiplasmodial activity, it is crucial that cost-effective simple procedures be conserved so that large-scale production and distribution to impoverished nations remains possible.
Following the validation and testing of current analogs, additional analogs may be evaluated based on informed decisions of the potential advantages of specific chemical groups and the success of similar moieties. Once a promising lead has been identified and validated, the mode of action and how the drug acts to mediate host parasite-interactions on a molecular level may be researched and the drug tailored for that specific functionality. Mechanisms of action have already been postulated for related arylpyrroles, informing the choice of analogs and pursuit of specific lead compounds.[9]
Open source science has contributed significantly to the rapid identification of promising drug leads. Contributions from a variety of fields of expertise allow both synthetic and biological concerns for these lead compounds to be addressed, giving a more holistic view of the nature of drug design goals. Stimulating discussion and participation, this free exchange of ideas enables involvement of the wider scientific community, accelerating research and development so that it may be possible to find an effective solution to a devastating problem as soon as possible.
References
- Wells, T.N.C., Alonso, P.L.and Gutteridge, W.E. “New Medicines to improve control and contribute to the eradication of Malara”, Nature Reviews,2009, Vol. 8, pp879-891
- Panosian, CB. “Economic access to effective drugs for falciparum malaria”, Clinical Infectious Diseases, 2005, Vol. 40, no. 5, pp. 713-717
- Wells T.N.C “Is the tide turning for New Malaria Medicines”, Science, 2010, Vol. 329 pp.1153-1154
- Francisco-Javier, G., Sanz, LM., Jaume, V. “Thousands of chemical starting points for antimalarial lead identification”, Nature, 2010, Vol. 465, no. 7296, pp305-U56
- Calderon, F., Barros, D., Beuno Jose, M. et al. “An Invitation to Open Innovation in Malaria Drug Discovery: 47 Quality Starting Points from the TCAMS”, ACS Chemistry Letters, 2011, Vol.2, no.10, pp741-746
- The Synaptic Leap – Malaria Research Community (Updated 2011)
- Todd, M., Ylioja, P. (2011). Open Source Drug Discovery – Malaria. Open wet ware.
- Lab Blog: Malaria.ourexperiment.org blog (Updated 2011)
- Lee, BH., Lee, MJ., Park, S,. Oh, DC,. Elsasser, S,. Chen, PC,. Gartner, C,. Dimova, N,. Hanna, J,. Gygi, SP,. Wilson, SM,. King, RW,. Finley, D. “Enhancement of proteasome activity by a small-molecule inhibitor of USP14”, Nature, 2010, Vol. 467, pp179-184