
PALINAVIR, BILA-2011-BS
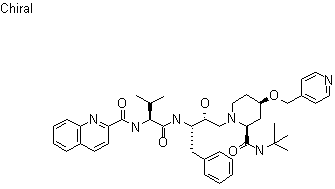
UNII-632S1WU9Z2, 154612-39-2, n-[(1s)-1-[[(1s,2r)-1-benzyl-3-[(2s,4r)-2-(tert-butylcarbamoyl)-4-(4-pyridylmethoxy)piperidino]-2-hydroxypropyl]carbamoyl]-2-methylpropyl]quinaldamide,
N-[(2S)-1-[[(2S,3R)-4-[(2S,4R)-2-(tert-butylcarbamoyl)-4-(pyridin-4-ylmethoxy)piperidin-1-yl]-3-hydroxy-1-phenylbutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]quinoline-2-carboxamide
Molecular Formula:C41H52N6O5
Molecular Weight:708.88878 g/mol
| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| Substituted pipecolinic acid derivatives as HIV protease inhibitors [US5614533] | 1997-03-25 | |
| Substituted pipecolinic acid derivatives as HIV protease inhibitors. [EP0560268] | 1993-09-15 | 1995-01-04 |
............................
PATENT
Scheme 5: Synthesis of Palinavir (6):

The organic solvent mentioned according to the invention is selected from the group consisting of organic solvents, wherein the organic solvents are polar aprotic such as DCM, THF, Ethyl acetate, acetone, DMF, acetonitrile, DMSO ; polar protic solvents such as lower alcohol particularly (C1-C6) alkyl alcohol, water, acetic acid ; non-polar solvents such as hexane, benzene, toluene, chloroform, pet. ether, 1,4-dioxane, heptane either alone or mixtures thereof . Additionally the purification or separation of crude product can be accomplished by known techniques viz. extraction, column chromatography in a suitable organic solvent with the aid of instruments such as TLC, HPLC, GC, mass spectroscopy, or distillation, crystallization, derivatization.
...............................
J Org Chem 1997,62(11),3440
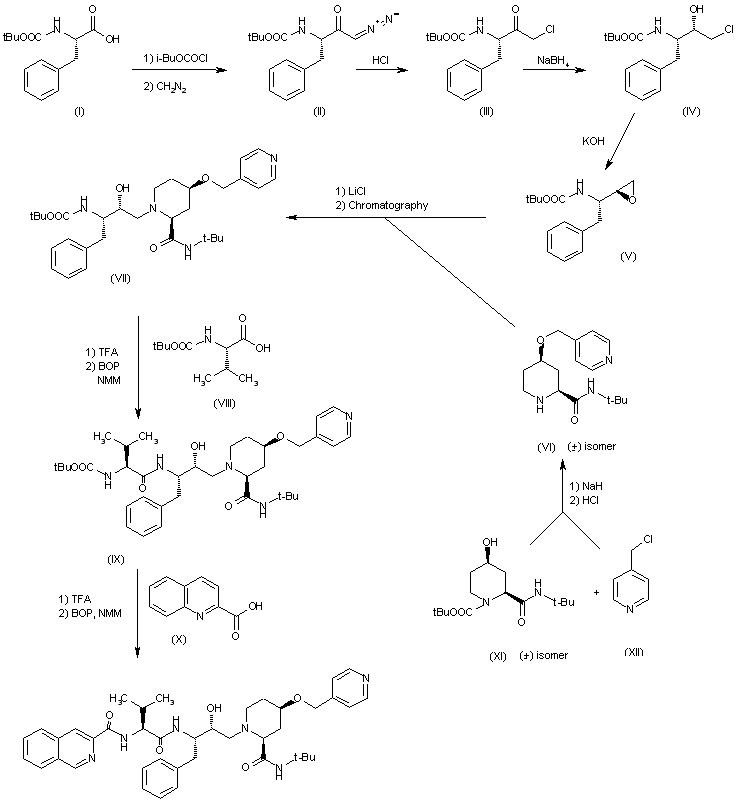
The reaction of tert-butoxycarbonyl-L-phenylalanine (I) with isobutyl chloroformate in THF gives the expected mixed anhydride which is treated with diazomethane and HCl yielding the corresponding chloromethyl ketone (II). The reduction of (II) with NaBH4 in THF affords the (S)-chlorohydrin (IV), which is treated with KOH in ethanol to obtain the chiral epoxide (V)(1,2). Ring opening of (V) with (?(cis)-N-tert-butyl-4-(4-pyridylmethoxy)piperidine-2-carboxamide (VI) by a treatment with LiCl in refluxing ethanol gives a mixture of diastereomers that is separated by chromatography giving the pure isomer (VII). The reaction of (VII) with tert-butoxycarbonyl-L-valine (VIII) by treatment first with trifluoroacetic acid (TFA), and condesation by means of BOP ((benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate) and NMM (N-methylmorpholine) affords the expected condensation product (IX). Finally, this compound is condensed with quinoline-2-carboxylic acid (X) by means of BOP and NMM as before. 2) The piperidine (VI) has been obtained by condensation of (?(cis)-N-(tert-butoxycarbonyl)-4-hydroxypiperidine-2-carboxamide (XI) with 4-(chloromethyl)pyridine (XII) by means of NaH in DMS, followed by hydrolysis with HCl.
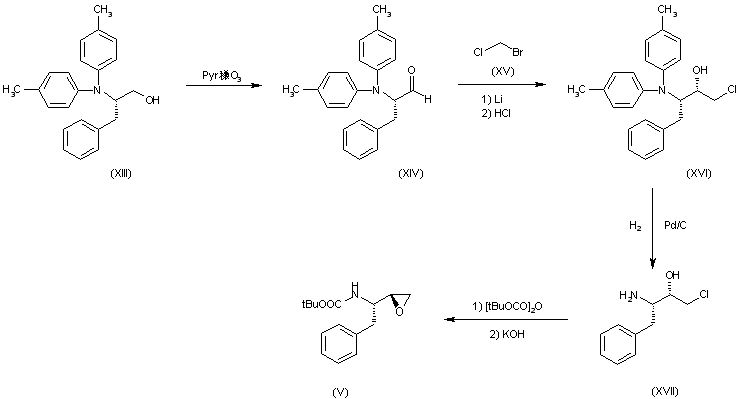
Palinavir can also be obtained as follows: The controlled oxidation of 2(S)-(dibenzylamino)-3-phenyl-1-propanol (XIII) with pyridine-SO3 complex in DMSO gives the corresponding aldehyde (XIV), which is condensed with bromochloromethane (XV) by means of Li in THF followed by hydrolysis with HCl yielding regioselectively the 1-chloro-2-butanol (XVI). The debenzylation of (XVI) by hydrogenation over Pd/C affords the free amine (XVII), which is treated with tert-butoxycarbonyl anhydride/triethylamine and dehydrochlorinated with KOH in methanol to give the desired chiral epoxide (V).
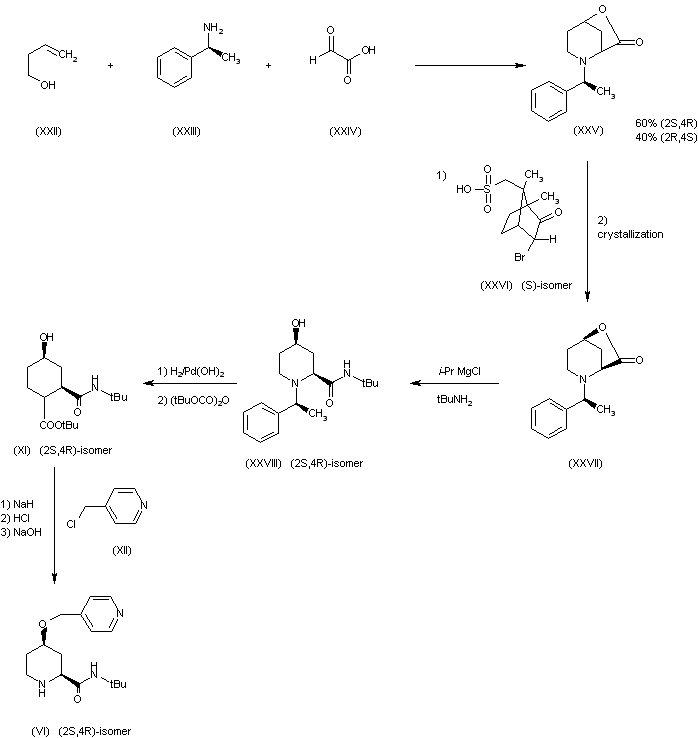
The chiral piperidine (2S,4R)(VI) has been obtained as follows: The cyclization of 3-buten-1-ol (XXII) with (S)-1-phenylethylamine (XXIII) and glyoxylic acid (XXIV) by means of tosyl chloride in THF gives a mixture of the (2S,4R) and (2R,4S) lactones (XXV), which is resolved by fractional crystallyzation of their salts with the chiral camphorsulfonic acid (XXVI), followed by elimination of the acid with ammonia to afford (2S,4R)(XXVII). The reaction of lactone (XXVII) with isopropylmagnesium chloride and tert-butylamine in THF gives (2S,4R)-N-tert-butyl-4-hydroxy-1-(1(S)-phenylethyl)piperidine-2-carboxamide (XXVIII), which is debenzylated by hydrogenation and protected with tert-butoxycarbonyl anhydride yielding (2S,4R)-N-(tert-butoxycarbonyl)-4-hydroxypiperidine-2-carboxamide (2S,4R)(XI), which is finally condensed with 4-(chloromethyl)pyridine (XII) as before to obtain the chiral piperidine (2S,4R)(VI), already reported.
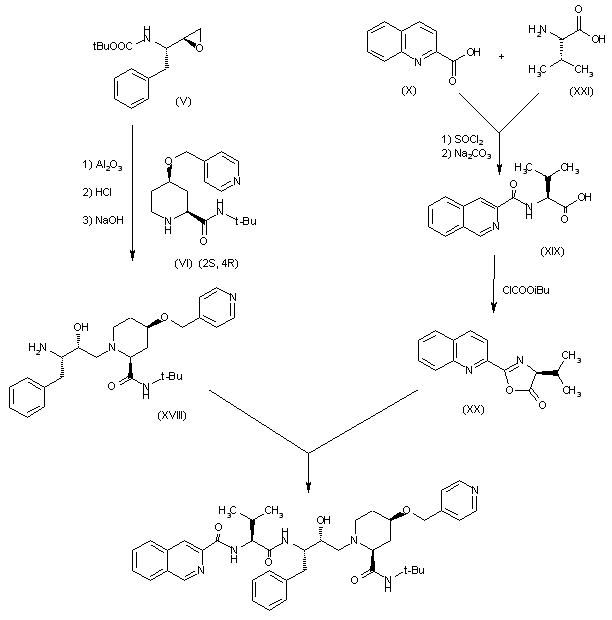
The condendsation of epoxide (V) with (2S,4R)(VI) by means of basic alumina in THF, followed by elimination of the protecting group with HCl and NaOH yields directly the condensation product (XVIII) as a pure diastereomer and with a free amino group. Finally, this compound is condensed with N-(2-quinolylcarbonyl)-L-valine (XIX) through its activation compound with isobutyl chloroformate (the 4(S)-isopropyl-2-(2-quinolyl)oxazol-5(4H)-one (XX)). The N-acyl-L-valine (XIX) has been obtained by acylation of L-valine (XXI) with quinoline-2-carboxylic acid (X) through its acyl chloride obtained with SOCl2.
.............................

Palinavir is an inhibitor with five chiral centers. It contains the amino acid valine and pipecolinin acid. The previous way to create this drug faced three major obstacles. First, the reaction from 2 to 3 used diazomethane. Therefore, is is difficult, if not impossible, to produce large quantities. Secondly, the steps included in going from 4 to 5 gave way to racemers which is very inefficient. Finally, chromatography is needed at two separate times.

Four issues were addresses in route to product 1. First, because of the number of chiral centers, stereochemical control was a concern. high chemical yields were a second concern. Also, multi step procedures were advantageous to cut down on purification steps. Finally, the synthesis tried to restrict the use of hazardous reagents. The following retrosynthesis reaction was conceived and three target molecules were identified as seen in figure 1.

Molecule 3 uses a diaseteroselective addition of in situ (chloromethyl)lithium to N,N-dibenzylphenylalaninol and is derived from a four step process.

Recrystallization of 13 is required. Molecule 14 was not reached because it posed a problem later in the reaction. The N-benzyl protection group could not be removed to react with 9.

8 is a derivative of naturally occurring pipocolic acid, 16, named 3-buten-1-ol. Selective crystallization of diastereomeric salts can lead to 17a, but a more efficient way is by having a 60:40 mixture of lactones 17a,b. This leads to 18a,b using a Brodroux process. Crystallization of 18a,b lead to a poor overall yield. Instead, 18a,b undergoes salt crystallization with (-)-camphorsulfonic acid. Finally, 18a underwent hydrolysis and then addition of di-tert butyl dicarbonate leads to 8.
8 was then transformed to 5 in a three step process.
 8 was added to NaOH and alkylated with 4-picolyl chloride. The protecting group was lost with the addition of acid.
8 was added to NaOH and alkylated with 4-picolyl chloride. The protecting group was lost with the addition of acid.  Derivation of 9 was started by a simple substitution of 19, quinoline-2-carboxylic acid, to 20, an acid chloride, with the help of thionyl chloride. Acylation of amino acid L-valine to 20 was accomplished by a biphasic system.
Derivation of 9 was started by a simple substitution of 19, quinoline-2-carboxylic acid, to 20, an acid chloride, with the help of thionyl chloride. Acylation of amino acid L-valine to 20 was accomplished by a biphasic system. In the original synthesis of palinavir, a 2:1 mixture of 3 to 5 was needed to produce only ~35% of 6 and flash chromatography was needed. On a large scale without chromatography, 6 was produced with a 85% yield, but 21 was also produced. To keep the production of 21 to a minimum, the reaction was performed in a solution that was degassed. This insured that the pyridine ring would not react in the presence of air. With this precaution, only 1-2% of the yield was 21. A washing of the solution with 1 M KH2PO4 removed and left over 5. Deprotection was achieved with the addition of concentrated HCl and followed by adding NaOH. The product of 10 was a "viscous syrup". 22 was 1-1.5% of the product and was not removed before the addition of 9 to form 80-85% palinavir.
In the original synthesis of palinavir, a 2:1 mixture of 3 to 5 was needed to produce only ~35% of 6 and flash chromatography was needed. On a large scale without chromatography, 6 was produced with a 85% yield, but 21 was also produced. To keep the production of 21 to a minimum, the reaction was performed in a solution that was degassed. This insured that the pyridine ring would not react in the presence of air. With this precaution, only 1-2% of the yield was 21. A washing of the solution with 1 M KH2PO4 removed and left over 5. Deprotection was achieved with the addition of concentrated HCl and followed by adding NaOH. The product of 10 was a "viscous syrup". 22 was 1-1.5% of the product and was not removed before the addition of 9 to form 80-85% palinavir.
Coupling of 10 and 9 is the final step in the synthesis , although there are still some purification steps left.

Two recrystallizations were required for the final 99.6% purity.

.............................
J. Org. Chem., 1997, 62 (11), pp 3440–3448
DOI: 10.1021/jo9702655

Palinavir is a potent peptidomimetic-based HIV protease inhibitor. We have developed a highly convergent and stereoselective synthesis which is amenable to the preparation of multikilogram quantities of this compound. The synthetic sequence proceeds in 24 distinct chemical steps (with several integrated, multistep operations) from commercially available starting materials. No chromatographies are required throughout the process, and the final product is purified by crystallization of its dihydrochloride salt to >99% homogeneity.
crude palinavir (1) as a thick brown oil (yield not determined). HPLC analysis (Supelcosil LZ-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): 1, tR 17.80 min (84.1%); 24, tR 18.47 min (2.0%); 25, tR 19.97 min (1.45%).
palinavir dihydrochloride (1750 g, 51% yield) containing 0.25% w/w isopropanol (by 1H NMR):
mp 175−185 °C.
[α]25D −13.0° (c 1, MeOH). [α]25Hg365 +44.9° (c 1, MeOH).
IR (KBr) ν 3700−2300, 1660, 1555, 1520 cm-1.
1H NMR (DMSO-d6) δ 10.00 (broad s, 1H), 8.88 (d, J = 6.3 Hz, 2H), 8.61 (d, J = 8.4 Hz, 1H), 8.60 (s, 1H), 8.51 (d, J = 9.6 Hz, 1H), 8.35 (d, J = 8.7 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 7.94 (d, J = 6.0 Hz, 2H), 7.89 (t, J = 7.6 Hz, 1H), 7.74 (t, J = 7.5 Hz, 1H), 7.19 (d, J = 7.2 Hz, 2H), 7.08 (t, J = 7.5 Hz, 2H), 6.91 (t, J = 7.3 Hz, 1H), 4.86 (AB quartet, 2H), 4.37 (broad t, J = 7.8 Hz, 1H), 4.21 (d, J = 11.4 Hz, 1H), 4.11 (broad m, 1H), 3.96 (broad m, 1H), 3.80−3.65 (m, 2H), 3.26 (t, J = 7.4 Hz, 1H), 3.15−3.01 (m, 2H), 2.94 (broad d, J = 12.0 Hz, 1H), 2.62 (dd, J = 13.6, 10.6 Hz, 1H), 2.56 ((broad d, J = 12.0 Hz, 1H), 2.20−2.05 (m, 2H), 1.86 (m, 1H), 1.69 (q, J = 11.7 Hz, 1H), 1.31 (s, 9H), 0.81 (d, J = 6.3 Hz, 3H), 0.80 (d, J = 6.6 Hz, 3H).
13C NMR (DMSO-d6) δ 170.4, 166.4, 163.3, 158.3, 149.5, 145.9, 141.9, 138.6, 138.2, 130.7, 129.3, 129.1, 129.0, 128.3, 128.2, 128.0, 125.9, 124.1, 118.6, 72.3, 68.8, 67.2, 64.8, 58.0, 57.8, 54.4, 51.3, 51.1, 35.4, 34.1, 31.1, 28.2, 19.5, 17.9.
FAB-MS m/z 709 (MH+ of free base). Anal. Calcd for C41H54Cl2N6O5 (corrected for 8% water content as determined by Karl Fisher analysis and 0.25% w/w isopropanol as determined by 1H NMR): C, 58.31; H, 7.29; N, 9.93. Found: C, 57.76; H, 7.25; N, 9.89. Titration of HCl content using NaOH: 2.09 ± 0.03 mol HCl. HPLC homogeneity (Supelcosil LC-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): palinavir dihydrochloride, tR 18.24 min (99.51%); 25 tR 20.39 min (0.33%). HPLC homogeneity (Nova-Pak C8, 20−80% MeCN/50 mM NaH2PO4 in 25 min, 1 mL/min flow rate): palinavir dihydrochloride, tR 15.52 min (99.67%); 25 tR 13.52 min (0.33%).
PURE palinavir (1) as a white amorphous powder (1902 g, 84% yield):
mp 100−107 °C. [α]25D −11.5° (c 1, MeOH).
IR (KBr) ν 3700−3100, 1660, 1520, 1495 cm-1.
1H NMR (CDCl3) δ 8.54 (d, J = 5.7 Hz, 2H), 8.48 (d, J = 8.6 Hz, 1H), 8.31 (d, J = 8.6 Hz, 1H, part of AB), 8.22 (d, J = 8.3 Hz, 1H, part of AB), 8.13 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.80 (t, J = 7.6 Hz, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.25 (d, J = 5.4 Hz, 2H), 7.13 (d, J = 7.3 Hz, 2H), 7.07 (t, J = 7.5 Hz, 1H), 6.92 (t, J = 7.3 Hz, 1H), 6.59 (d, J = 8.3 Hz, 1H), 6.57 (s, 1H), 4.61 (d, J = 13.4 Hz, 1H, part of AB), 4.51 (d, J = 13.4 Hz, 1H, part of AB), 4.32 (dd, J = 8.6, 6.4 Hz, 1H), 4.22 (m, 1H), 3.97 (m, 1), 3.47−3.33 (m, 2H), 2.94 (dd, J = 14.3, 4.1 Hz, 1H), 2.89 (d, J= 8.6 Hz, 1H), 2.79−2.72 (m, 1H), 2.77 (dd, J = 14.3, 10.8 Hz, 1H), 2.43 (dd, J = 13.4, 8.3 Hz, 1H), 2.40−2.25 (m, 3H), 1.95 (broad d, J = 12.4 Hz, 1H), 1.65 (q J = 11.8 Hz, 2H), 1.32 (s, 9H), 0.95 (d, J = 7.0 Hz, 3H), 0.83 (d, J = 6.7 Hz, 3H).
13C NMR (CDCl3) δ 171.6, 171.2, 165.0, 149.8, 148.8, 147.9, 146.5, 137.6, 137.5, 130.3, 129.9, 129.5, 129.4, 129.0, 128.8, 128.5, 128.2, 127.7, 126.4, 121.7, 118.8, 75.0, 71.9, 68.1, 66.7, 59.4, 56.9, 54.6, 50.9, 50.2, 34.8, 33.3, 29.8, 29.7, 28.7, 19.6, 17.5.
FAB-MS m/z 709 (MH+). Anal. Calcd for C41H52N6O5(corrected for 0.7% water content as determined by Karl Fisher analysis): C, 68.98; H, 7.42; N, 11.77. Found: C, 68.71; H, 7.47; N, 11.71. HPLC homogeneity (Supelcosil LC-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): palinavir (1), tR 17.83 min (99.59%); 25 tR20.00 min (0.41%). HPLC homogeneity (Nova-Pak C8, 10−80% MeCN/50 mM NaH2PO4 in 25 min, 1 mL/min flow rate): palinavir (1), tR 17.37 min (99.51%); 25 tR 15.87 min (0.49%).
| REFERENCE | ||
|---|---|---|
| 1 | * | ARUN K. GHOSH ET AL: "The Development of Cyclic Sulfolanes as Novel and High-Affinity P2 Ligands for HIV-1 Protease Inhibitors", JOURNAL OF MEDICINAL CHEMISTRY, vol. 37, no. 8, 1 April 1994 (1994-04-01), pages 1177-1188, XP055057710, ISSN: 0022-2623, DOI: 10.1021/jm00034a016 |
| 2 | * | KAY BRICKMANN ET AL: "Synthesis of Conformationally Restricted Mimetics of [gamma]-Turns and Incorporation into Desmopressin, an Analogue of the Peptide Hormone Vasopressin", CHEMISTRY - A EUROPEAN JOURNAL, vol. 5, no. 8, 2 August 1999 (1999-08-02), pages 2241-2253, XP055057517, ISSN: 0947-6539, DOI: 10.1002/(SICI)1521-3765(19990802)5:8<2241: :AID-CHEM2241>3.0.CO;2-L |
| 3 | * | KIRAN I N C ET AL: "A concise enantioselective synthesis of (+)-goniodiol and (+)-8-methoxygoniodiol via Co-catalyzed HKR of anti-(2SR, 3RS)-3-methoxy-3-phenyl-1, 2-epoxypropane", TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 52, no. 3, 19 January 2011 (2011-01-19), pages 438-440, XP027558447, ISSN: 0040-4039 [retrieved on 2010-12-14] |
| 4 | * | M. TOKUNAGA: "Asymmetric Catalysis with Water: Efficient Kinetic Resolution of Terminal Epoxides by Means of Catalytic Hydrolysis", SCIENCE, vol. 277, no. 5328, 15 August 1997 (1997-08-15), pages 936-938, XP055057541, ISSN: 0036-8075, DOI: 10.1126/science.277.5328.936 |
| 5 | * | PARKES K E B ET AL: "STUDIES TOWARD THE LARGE-SCALE SYNTHESIS OF THE HIV PROTEINASE INHIBITOR RO 31-8959", JOURNAL OF ORGANIC CHEMISTRY, ACS, US, vol. 59, no. 13/16, 1 January 1994 (1994-01-01), pages 3656-3664, XP002011975, ISSN: 0022-3263, DOI: 10.1021/JO00092A026 |
| 6 | * | R. SANTHOSH REDDY ET AL: "Co(iii)(salen)-catalyzed HKR of two stereocentered alkoxy- and azido epoxides: a concise enantioselective synthesis of (S,S)-reboxetine and (+)-epi-cytoxazone", CHEMICAL COMMUNICATIONS, vol. 46, no. 27, 1 January 2010 (2010-01-01), page 5012, XP055057537, ISSN: 1359-7345, DOI: 10.1039/c0cc00650e |
| 7 | * | SHINJI NAGUMO ET AL: "Intramolecular Friedel-Crafts type reaction of vinyloxiranes linked to an ester group", TETRAHEDRON, vol. 65, no. 47, 1 November 2009 (2009-11-01), pages 9884-9896, XP055057655, ISSN: 0040-4020, DOI: 10.1016/j.tet.2009.09.037 |
| 8 | * | SUNITA K. GADAKH ET AL: "Enantioselective synthesis of HIV protease inhibitor amprenavir via Co-catalyzed HKR of 2-(1-azido-2-phenylethyl)oxirane", TETRAHEDRON: ASYMMETRY, vol. 23, no. 11-12, 1 June 2012 (2012-06-01), pages 898-903, XP055057475, ISSN: 0957-4166, DOI: 10.1016/j.tetasy.2012.06.003 |