
Decernotinib
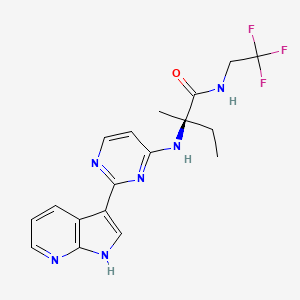
Decernotinib
N2-[2-(1H-Pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl]-N-(2,2,2-trifluoroethyl)-D-isovalinamide
(R)-2-(2-(lH-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-ylamino)-2-methyl-N-(2,2,2- trifluoroethyl)butanamide
Vertex Pharmaceuticals Inc
UNII-MZK2GP0RHK, VX-509, VRT-831509, cas 944842-54-0
Molecular Formula: C18H19F3N6O
Molecular Weight: 392.37827
In phase 3 for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis.
 DECERNOTINIB
DECERNOTINIB
The Janus kinases (JAK) are a family of tyrosine kinases consisting of JAK1, JAK2, JAK3, and TYK2. The JAKs play a critical role in cytokine signaling. The down-stream substrates of the JAK family of kinases include the signal transducer and activator of transcription (STAT) proteins. JAK/STAT signaling has been implicated in the mediation of many abnormal immune responses such as psoriasis. Moreover, JAK kinases represent an established therapeutic target for this disease.
For example, JAK kinases are an established therapeutic target for treating psoriasis. Stump K. L., et al., Arthritis Res. Ther. (201 1) 13:R68; Fridman J.S., et al., J Immunol. (2010) 184:5298-5307; West K., Curr. Op. Investig. Drugs (2009) 10:491-504; Kremer J. M. et al., Arthritis Rheumatism (2009) 60(7):1895- 1905; Xiong, W. et al., Ther Adv Musculoskelet Dis. (201 1) 3(5): 255-266; Panes, J. et al. 19th Ann. Eur. Gastroenterology Week (Oct 22-26, 2011) Stockholm, SE, PI 456; and Drugs in R & D "Tofacitinib" (2010) 10(4):271-84.
Compounds described as kinase inhibitors, particularly the JAK family kinases, are disclosed in WO 2005/095400 and WO 2007/084557. Also disclosed in these publications are processes and intermediates for preparing these compounds
Decernotinib ( VX-509 ) is an oral selective JAK3 inhibitor being evaluated for the treatment of rheumatoid arthritis ( RA ). This was a 24-week, randomized, placebo-controlled, double-blind, phase 2 study of four dosing regimens of Decernotinib, administered to patients with RA with inadequate response to Methotrexate ( MTX ).
The aim of the study was to assess the efficacy and safety of four dosing regimens of VX-509 administered to patients with rheumatoid arthritis on stable background Methotrexate therapy.
Patients with active rheumatoid arthritis ( C-reactive protein [ CRP ] greater than ULN, greater than or equal to 6 swollen joints [ of 66 ], and greater than or equal to 6 tender joints [ of 68 ] ) taking stable doses of MTX were randomized 1:1:1:1:1 to receive placebo or one of four dosing regimens of Decernotinib ( 100 mg QD, 150 mg QD, 200 mg QD, or 100 mg BID ) for a duration of 24 weeks.
The primary efficacy endpoints at week 12 were met and have previously been reported; 24-week efficacy and safety results are now reported.
A total of 358 patients were randomized and received greater than or equal to 1 dose of study drug; 81% of patients were female, with a mean age of 53 years.
At baseline, the mean tender joint count was 23.8, the mean swollen joint count was 16.1, and the average disease duration was 7.3 years.
At baseline, the mean tender joint count was 23.8, the mean swollen joint count was 16.1, and the average disease duration was 7.3 years.
After 24 weeks of treatment the proportion of patients achieving ACR20, ACR50, ACR70, DAS28 ( CRP ) less than 2.6 and DAS28 ( ESR ) less than 2.6 and the decrease from baseline in DAS28 ( CRP ) were statistically significantly greater in each of the Decernotinib dose groups than in the placebo group.
Over 24 weeks, the percentage of patients with any adverse event was higher in the Decernotinib group ( all Decernotinib dose groups combined ) ( 59.9% ) relative to placebo ( 42.3% ) and led to study discontinuation in 9.1% and 8.5% of patients in the Decernotinib and placebo groups, respectively.
The most common adverse reactions in the Decernotinib group were headache ( 8.7% ), hypercholesterolemia ( 5.2% ), and diarrhea ( 4.5% ).
Serious adverse reactions occurred in similar proportions of patients receiving Decernotinib ( 7.3% ) or placebo ( 5.6% ), but there were more serious infections in the Decernotinib group ( 3.5% ) compared with placebo ( 1.4% ).
Through 24 weeks there were two serious adverse effects that resulted in death; one was cardiac failure in the Decernotinib 100 mg BID group ( previously reported ) and one was pancytopenia in a patient with pneumonia in the Decernotinib 200 mg QD group.
Elevations in transaminase levels and decreases in median neutrophil and lymphocyte counts were observed in the Decernotinib groups and were generally mild.
The most common adverse reactions in the Decernotinib group were headache ( 8.7% ), hypercholesterolemia ( 5.2% ), and diarrhea ( 4.5% ).
Serious adverse reactions occurred in similar proportions of patients receiving Decernotinib ( 7.3% ) or placebo ( 5.6% ), but there were more serious infections in the Decernotinib group ( 3.5% ) compared with placebo ( 1.4% ).
Through 24 weeks there were two serious adverse effects that resulted in death; one was cardiac failure in the Decernotinib 100 mg BID group ( previously reported ) and one was pancytopenia in a patient with pneumonia in the Decernotinib 200 mg QD group.
Elevations in transaminase levels and decreases in median neutrophil and lymphocyte counts were observed in the Decernotinib groups and were generally mild.
Safety profiles were comparable across groups receiving Decernotinib.
In conclusion, all tested doses of Decernotinib significantly improved signs and symptoms of rheumatoid arthritis versus placebo when administered in combination with stable background Methotrexate therapy for 24 weeks.
Decernotinib was associated with small increases in adverse reactions rates, serious infections, and mostly minor laboratory abnormalities. ( Xagena )
Decernotinib was associated with small increases in adverse reactions rates, serious infections, and mostly minor laboratory abnormalities. ( Xagena )
Source: EULAR Meeting - van Vollenhoven R et al, Ann Rheum Dis 2014;73(Suppl2)
see
WO 2007084557
http://www.google.com/patents/WO2007084557A2?cl=en
.............................................
WO 2013006634
http://www.google.com/patents/WO2013006634A2?cl=en

Formula I is:
The present invention provides a process for preparing (R)-2-(2-(lH-pyrrolo[2,3- b]pyridin-3-yl)pyrimidin-4-ylamino)-2-methyl-N-(2,2,2-trifluoroethyl)butanamide of Formula la:
 la
la
comprising the steps of:
ivb) reacting lH-pyrrolo[2,3-b]pyridine (5a) with p-toluenesulfonyl chloride in the presence of an organic solvent to generate l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a)
 5a 9a
5a 9a
vb) reacting l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a) in an organic solvent with N-bromosuccinimide to generate 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a)
 vi) reacting 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a) with triisopropyl borate in the presence of a strong lithium base in an organic solvent to generate
vi) reacting 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a) with triisopropyl borate in the presence of a strong lithium base in an organic solvent to generate
l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) 0H
 8a
8a
vii) esterifying l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) with pinacolate alcohol in an organic solvent to generate
3 -(4,4,5 ,5 -tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -tosyl- 1 H-pyrrolo[2,3 -bjpyridine (la) :
 viiib) reacting 2,4-dichloropyrimidine (11a) with a hydrochloride salt of D-isovaline (15a) under coupling condition to generate a compound of Formula 2a
viiib) reacting 2,4-dichloropyrimidine (11a) with a hydrochloride salt of D-isovaline (15a) under coupling condition to generate a compound of Formula 2a
 11a 2a
11a 2a
ixb) reacting the compound of Formula 2a with HC1 to generate the hydrochloride salt of the compound of Formula 2a;
i) reacting the compound of Formula la with the compound of Formula 2a with in the presence of water, an organic solvent, an inorganic base, and a transition metal catalyst to generate a compound of Formula 3a,
 ii) deprotecting the compound of Formula 3a under basic conditions to generate a compound of Formula 4a
ii) deprotecting the compound of Formula 3a under basic conditions to generate a compound of Formula 4a
 4a ; and iii) reacting the compound of Formula 4a with 2,2,2-trifluoroethylamine in the presence of a coupling agent and an organic solvent to generate the compound of Formula la.
4a ; and iii) reacting the compound of Formula 4a with 2,2,2-trifluoroethylamine in the presence of a coupling agent and an organic solvent to generate the compound of Formula la.

 - l13C4, 15N2]
- l13C4, 15N2]


comprising the steps of:
ivb) reacting lH-pyrrolo[2,3-b]pyridine (5a) with p-toluenesulfonyl chloride in the presence of an organic solvent to generate l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a)
vb) reacting l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a) in an organic solvent with N-bromosuccinimide to generate 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a)
l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) 0H
vii) esterifying l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) with pinacolate alcohol in an organic solvent to generate
3 -(4,4,5 ,5 -tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -tosyl- 1 H-pyrrolo[2,3 -bjpyridine (la) :
ixb) reacting the compound of Formula 2a with HC1 to generate the hydrochloride salt of the compound of Formula 2a;
i) reacting the compound of Formula la with the compound of Formula 2a with in the presence of water, an organic solvent, an inorganic base, and a transition metal catalyst to generate a compound of Formula 3a,
.........................................................................
WO 2013070606
http://www.google.com/patents/WO2013070606A1?cl=en
..........................................................
patent WO2014074471
WO2014074471 claiming use of heterocyclic compound (preferably decernotinib) for treating psoriasis. Vertex is developing decernotinib, an oral JAK 3 inhibitor, for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis. As of July 2014, the drug is Phase 3 trials.
http://www.google.com/patents/WO2014074471A1?cl=en
Table 1:
COMPD 1 IS DECERNOTINIB


Example 1: Analytical Methods Used
[0260] (A) HPLC on C18 column. Mobile phase was acetonitrile/water/TFA (60:40:0.1). Flow rate was 1.0 mL/min. Detection at wavelength of 230 nm. Run time was 25-26 minutes.
[0261] (B) HPLC on C18 column. Mobile phase was acetonitrile/water/TFA (90: 10:0.1). Flow rate was 1.0 mL/min. Detection at wavelength of 230 nm.
[0262] (C) HPLC on a Waters XBridge Phenyl column, 4.6 x 150 mm, 3.5 μπι. Mobile phase A was water/1 M ammonium formate, pH 4.0 (99: 1). Mobile phase B was
acetonitrile/water/ 1M ammonium formate, pH 4.0 (90:9:1). Gradient 5 % to 90 % B in 15 minutes. Total run time 22 minutes. Flow rate 1.5 mL/min. Detection at UV, 245 nm.
T = 25 °C.
[0263] (D) HPLC on a Waters XBridge Phenyl column, 4.6 x 150 mm, 3.5 μπι. Mobile phase A was water/1 M ammonium formate, pH 4.0 (99: 1). Mobile phase B was
acetonitrile/water/ 1M ammonium formate, pH 4.0 (90:9: 1). Gradient 15% to 90 % B in 15 minutes. Total run time 22 minutes. Flow rate 1.5 mL/min. Detection at UV, 220 nm.
T = 35 °C.
[0264] Example 2: Preparation of Compounds of Formula I [0265] General Synthetic Scheme

[0266] The Boc-protected amino acid starting material (1) undergoes amidation in the presence of an activating agent, a coupling reagent, and the acid salt of the amine HNR7R17 to generate the Boc-protected amide intermediate (2). The amide intermediate (2) is
deprotected under acidic conditions and reacted with the halogenated heteroaryl (3) to generate the aminoheteroaryl intermediate (4). Boronated azaindole (5) is coupled with the aminoheteroaryl intermediate (4) under cross-coupling condition to generate the compound of Formula I.
.....................................................................................
Patent
http://www.google.com/patents/US8163917
| 346 | M+H393.20 | RT 1.60 | (DMSO-d6, 300 MHz) 11.95 (bs, 1H), 8.7 (d, |
| 1H), 8.25 (m, 2H), 8.12 (d, 1H), 8.02 (d, 1H), | |||
| 7.28 (s, 1H), 7.13 (dd, 1H), 6.38 (bd, 1H), 3.75 | |||
| (m, 2H), 2.06 (m, 1H), 1.83 (m, 1H), 1.46 (s, | |||
| 3H), 0.8 (t, 3H); |

| 346 |
|
Example 1 Preparation of Compounds of the Invention
General Synthetic Scheme

Step 1
To a stirred solution of Boc-valine (1; R1 is Me; 3.8 g, 0.02 mol), EDC (4.63 g, 0.024 mol), HOBt (4.0 g, 0.026 mol), DIEA (10.5 mL, 0.06 mol) in 100 mL of DCM is added trifluoroethylamine HCl (2.92 g, 0.022 mol). The reaction mixture is stirred for 16 h. It is concentrated to dryness and redissolved in EtOAc, washed successively with 0.5N HCl, saturated aqueous solution of NaHCO3 and brine. The organic layer is dried (Na2SO4) and concentrated in vacuo to give 5.4 g (98%) of 2 as a white solid.
Step 2
Compound 2 (5.32 g, 0.0197 mol) is deprotected with a 1:1 mixture of DCM/TFA at rt for 45 min. Concentration to dryness gives the intermediate amine that is used directly for the next step. A mixture of 5-fluoro-2,4-dichloropyrimidine (3; R is F; 3.28 g, 0.0197 mol), the crude amine TFA salt (5.25 g, 0.0197 mol) and DIEA (10.27 mL, 0.059 mol) are stirred in isopropanol at rt for 16 h. The reaction mixture is concentrated in vacuo and redissolved in EtOAc, washed successively with 0.5N HCl, saturated aqueous solution of NaHCO3 and brine. The organic layer is dried (Na2SO4) and concentrated in vacuo to give a crude oil that is subjected to chromatography (50% EtOAc/50% hexanes) to yield the desired compound 4.
Step 3
A mixture of 5 (30 mg, 0.075 mmol; prepared according to WO 2005/095400), 4 (23 mg, 0.075 mmol), Pd (Ph3P)4 (9 mg, 0.0078 mmol) and sodium carbonate 2M (115 uL, 0.23 mmol) in 1 mL of DME is microwaved at 150° C. for 10 minutes. The reaction mixture is filtered through a short pad of silica gel with 30% EtOAc-70% hexanes as eluent to provide, after concentration to dryness, the crude intermediate that is used directly for the next step.
The crude intermediate is dissolved in 1 mL of dry methanol and 200 uL of sodium methoxide in methanol 25% was added. The reaction mixture is stirred at 60° C. for 1 h and quenched with 6N HCl (154 uL). The mixture is dried under a flow of nitrogen and purified by reverse phase HPLC (10-60 MeCN/water w/0.5% TFA) to provide the desired material of formula 6a.
Compounds of formulae 6b and 6c may be prepared in an analogous manner using the appropriate starting reagents. For instance, a compound of formula 6b may generally be made by substituting Cert-butyl 2-(2,2,2-trifluoroethylcarbamoyl)pyrrolidine-1-carboxylate for compound 1, while a compound of formula 6c may generally be made by substituting tert-butyl 2-(2,2,2-trifluoroethylcarbamoyl)propan-2-ylcarbamate for compound 1.
Example 2 Analytical Results
Tables 4, 5 and 6 below depicts exemplary 1H-NMR data (NMR) and liquid chromatographic mass spectral data, reported as mass plus proton (M+H), as determined by electrospray, and retention time (RT) for certain compounds of the present invention, wherein compound numbers in Tables 4, 5 and 6 correspond to the compounds depicted in Tables 1, 2 and 3, respectively (empty cells indicate that the test was not performed):
PATENTS
4-25-2012
|
Azaindoles Useful as Inhibitors of Janus Kinases
| |
8-4-2010
|
Azaindoles useful as inhibitors of janus kinases
|
new patent
WO-2014110259
| US8450489 * | Mar 1, 2012 | May 28, 2013 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| US8530489 * | May 22, 2012 | Sep 10, 2013 | Vertex Pharmaceuticals Incorporated | 5-cyano-4-(pyrrolo [2,3B] pyridine-3-yl)-pyrimidine derivatives useful as protein kinase inhibitors |
| US8686143 * | Oct 25, 2011 | Apr 1, 2014 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of Janus kinases |
| US20120157429 * | Oct 25, 2011 | Jun 21, 2012 | Wannamaker Marion W | Compounds useful as inhibitors of janus kinases |
| US20120165307 * | Mar 1, 2012 | Jun 28, 2012 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| US20120309963 * | May 22, 2012 | Dec 6, 2012 | Vertex Pharmaceuticals Incorporated | 5-cyano-4- (pyrrolo [2,3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| US20130237516 * | Apr 25, 2013 | Sep 12, 2013 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| WO2013173506A2 | May 15, 2013 | Nov 21, 2013 | Rigel Pharmaceuticals, Inc. | Method of treating muscular degradation |
| WO2005095400A1 | Mar 30, 2005 | Oct 13, 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 | Jan 17, 2007 | Jul 26, 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2013070606A1 * | Nov 6, 2012 | May 16, 2013 | Vertex Pharmaceuticals Incorporated | Methods for treating inflammatory diseases and pharmaceutical combinations useful therefor |