ote
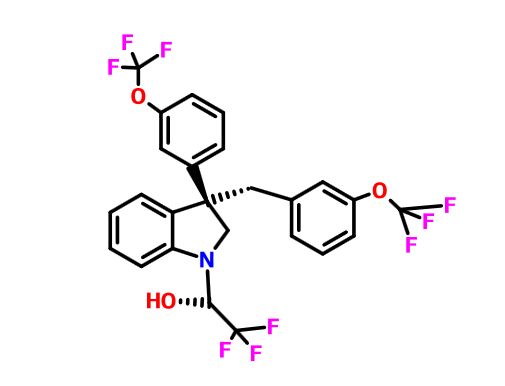
Indoline 7 as in ACS MEDCHEM LETTERS, DOI: 10.1021/acsmedchemlett.5b00404
and
eg 10 as in WO2015054088
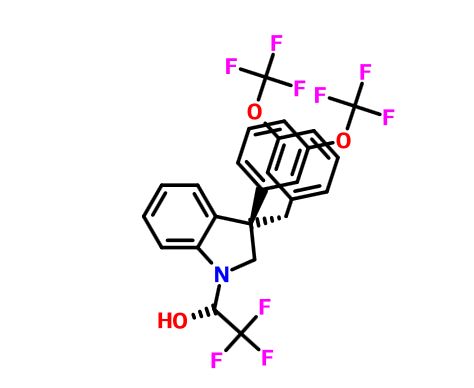
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
1H-Indole-1-ethanol, 2,3-dihydro-3-[3-(trifluoromethoxy)phenyl]-3-[[3- (trifluoromethoxy)phenyl]methyl]-α-(trifluoromethyl)-, (αR)-
cas 1699732-96-1 R ISOMER

see………….http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00404
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.5b00404/suppl_file/ml5b00404_si_001.pdf
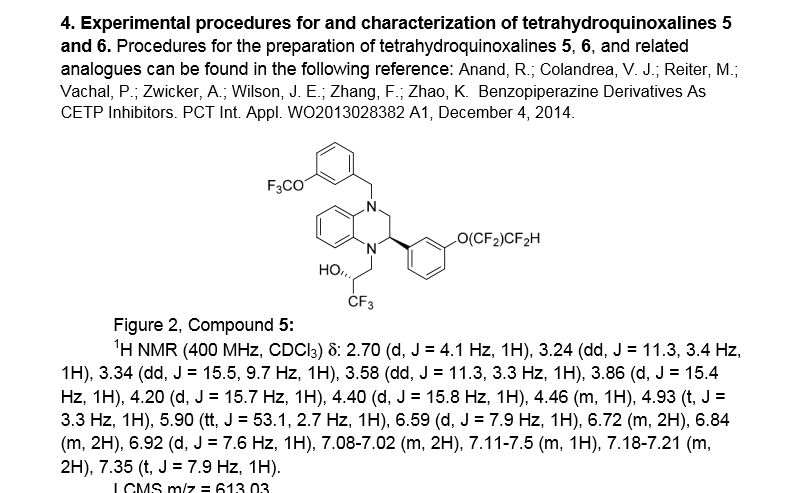 Example 18 as in patent
Example 18 as in patent

(R)- 1,1, 1 -trifluoro-3-((R)-4-(3-trifluoromethoxy)benzyl)-2-(3-(l, 1 ,2,2,-tetrafluoroethoxy)phenyl)-3,4- dihydroquinoxalin- 1 (2H)-yl)propan-2-ol
SPA: 15 nM
Example 18 was prepared from 2-bromo-l-(3-(l , 1 ,2,2,-tetrafluoroethoxy)phenyl)ethanone in three steps, using the reactions detailed in Schemes A6, A2 and Al . Spectral data are as follows: 1H NMR (400 MHz, CDC13) £2.70 (bd, J=4.1 Hz, IH), 3.24 (dd, J=l 1.3, 3.4 Hz, IH), 3.34 (dd, J=15.5, 9.7 Hz, IH), 3.58 (dd, J=l 1.3, 3.3 Hz, IH), 3.86 (d, J=15.4 Hz, IH), 4.20 (d, J=15.7 Hz, IH), 4.40 (d, J=15.8 Hz, IH), 4.46 (m, IH), 4.927 (t, J=3.3 Hz, IH), 5.90 (tt, J=53.1 , 2.7 Hz, IH), 6.59 (d, J= 7.9 Hz, IH), 6.72 (m, 2H), 6.84 (m, 2H), 6.92 (d, J=7.6 Hz, IH), 7.20 (m, 2H), 7.35 (t, J=7.9 Hz, IH), MS m/z = 613.03.
Scheme A12

Methyl 3 – { 1 – [(R)-3 ,3 ,3 -trifluoro-2-hy droxypropyl] -4- [3 -(trifluoromethoxy) benzyl]-l,2,3,4-tetrahydroquinoxalin-2-yl}benzoate (700 mg, 1.262 mmol) is made as described in Example 16 but with one stereochemical center unresolved. The compound was dissolved in MeOH (12.6mL), lithium hydroxide monohydrate (530 mg, 12.62 mmol) was added, and the reaction mixture was heated to 60°C for 4 hours. The crude mixture was dissolved in saturated ammonium chloride solution and extracted into EtOAc, the organic phase was dried with anhydrous magnesium sulfate, filtered, concentrated, and purified on a silica gel column with a 0-100% Hex/EtOAc gradient. The major peak was concentrated to afford 3-{l-[(R)-3,3,3-trifluoro-2-hydroxypropyl]-4-[3-(trifluoromethoxy)benzyl]-l,2,3,4-tetra-hydroquinoxalin-2-yl} benzoic acid. MS m/z = 541.09.

Patent
WO2015054088
http://google.com/patents/WO2015054088A1?cl=en
Scheme Al

Scheme A2

Scheme A3

R = Ar, NR2l C02R, CN, S02Me
es
es

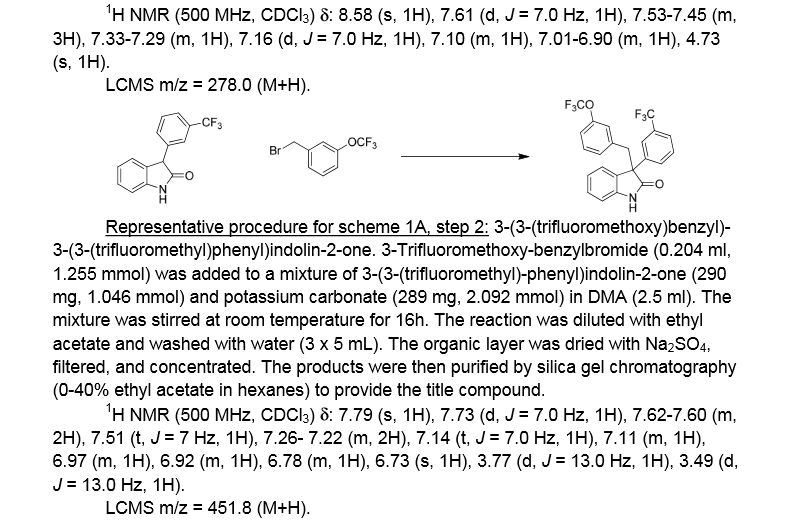
Example 1. (2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. This material was prepared according to Scheme Al, as described below.

3-(3-(trifluoromethyl)phenyl)indolin-2-one. Oxindole (1.598 g, 12 mmol), 3-bromo-a,a,a-trifluoromethyltoluene (2.009 ml, 14.40 mmol), potassium carbonate (3.32 g, 24.00 mmol), Pd2dba3 (0.220 g, 0.240 mmol), and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (0.458 g, 0.960 mmol) were combined in THF (12 ml) and the mixture was degassed with nitrogen. The solution was then heated to 80 °C for 18h. The mixture was cooled to room temperature, filtered through silica eluting with ethyl acetate, and concentrated. The material was then purified by silica gel chromatography (Biotage lOOg SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethyl)phenyl)indolin-2-one as a white solid.
1H NMR (500 MHz) δ 8.58 (s, 1H), 7.61 (d, J=7 Hz, 1H), 7.53-7.45 (m, 3H), 7.33-7.29 (m, 1H), 7.16 (d, J=7 Hz, 1H), 7.10 (m, 1H), 7.01-6.90 (m, 1H), 4.73 (s, 1H).

3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin-2-one . 3 -Trifluoromethoxy-benzylbromide (0.204 ml, 1.255 mmol) was added to a mixture of 3-(3-(trifluoromethyl)-phenyl)indolin-2-one (290 mg, 1.046 mmol) and potassium carbonate (289 mg, 2.092 mmol) (sodium carbonate may be used in place of potassium carbonate) in DMA (2.5 ml). The mixture was stirred at r.t. for 16h. The reaction was diluted with ethyl acetate and washed with water (3×5 mL). The organic layer was dried with Na2S04, filtered, and concentrated. The products were then purified by silica gel chromatography (Biotage 50g SNAP cartridge; 0-40%> ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-2-one .
1H NMR (500 MHz) δ 7.79 (s, 1H), 7.73 (d, J=7 Hz, 1H), 7.62-7.60 (m, 2H), 7.51 (t, J=7 Hz, 1H), 7.26- 7.22 (m, 2H), 7.14 (t, J=7.0 Hz, 1H), 7.11 (m, 1H), 6.97 (m, 1H), 6.92 (m, 1H), 6.78 (m, 1H), 6.73 (s, 1H), 3.77 (d, J=13 Hz, 1H), 3.49 (d, J=13 Hz, 1H).
LCMS m/z = 451.8 (M+H)

3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. Borane tetrahydrofuran complex (1.673 ml, 1.673 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indolin-2-one (302 mg, 0.669 mmol) in THF (1.5 ml). The mixture was heated to 70 °C for 20h. The reaction was cooled to room temperature and quenched with saturated NH4C1 solution, and this mixture was stirred vigorously for 20 minutes. The product was extracted with ethyl acetate. The extracts were dried over Na2S04, filtered, and concentrated. The product was purified by silica gel chromatography (Biotage 25g SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. This material may also be used without purification in the final step of the sequence, epoxide opening.
1H NMR (500 MHz) δ 7.66 (s, IH), 7.59 (d, J=7 Hz, IH), 7.53 (d, J=7 Hz, IH), 7.45 (t, J=8 Hz, IH), 7.18-7.13 (m, 2H), 7.04 (d, J=8 Hz, IH), 6.98 (d, J=7 Hz, IH), 6.81 (t, J=7.5 Hz, IH), 6.71 (m, 2H), 6.60 (s, IH), 3.83 (m, IH), 3.75-3.73 (m, 2H), 3.46 (d, J=13 Hz, IH), 3.41 (d, J=13 Hz, IH).
= 437.9 (M+H)

(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. (S)-2-(trifluoromethyl)oxirane (81 μΐ, 0.933 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline (136 mg, 0.311 mmol) in l,l,l,3,3,3-hexafluoro-2-propanol (412 μΐ, 3.91 mmol). The reaction was stirred at room temperature overnight. The solvent was removed and the product was purified by silica gel chromatography (Biotage 25 g SNAP cartridge; 0-25% ethyl acetate in hexanes) to provide (2R)- 1 ,1,1 -trifluoro-3 -(3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin- 1 -yl)propan-2-ol.
1H NMR (500 MHz) (mixture of diastereomers) δ 7.72 (s, 0.5 H), 7.69 (s, 0.5 H), 7.65 (d, J=6.5 Hz, 0.5 H), 7.61 (d, J=7.5 Hz, 0.5 H), 7.56 (s, 1H), 7.50 (m, 1H), 7.25-7.17 (m, 2H), 7.07 (broad s, 2H), 6.91-6.89 (m, 1H), 6.79-6.75 (m, 1H), 6.53 (m, 2H), 4.00 (broad s, 1H), 3.83 (d, J= 9 Hz, 0.5H), 3.77 (d, J=9 Hz, 0.5H), 3.59-3.55 (m, 1H), 3.45-3.43 (m, 1H), 3.39-3.29 (m, 2H), 3.21-3.15 (m, 1H), 2.32 (m, 0.5H), 2.15 (m, 0.5H).
LCMS m/z = 549.8 (M+H)
Examples 1-25, in the table below, were prepared according to Scheme Al in a

ABOUT AUTHOR
 https://www.linkedin.com/in/jonathan-wilson-23206523
https://www.linkedin.com/in/jonathan-wilson-23206523
October 2013 – Present (2 years 4 months)
May 2009 – October 2013 (4 years 6 months)
October 2007 – May 2009 (1 year 8 months)
2000 – 2002 (2 years)
///////CETP inhibition, cholesterol ester transfer protein, HDL, indoline, tetrahydroquinoxaline, merck, discovery
c21ccccc1N(C[C@@]2(c3cccc(c3)OC(F)(F)F)Cc4cc(ccc4)OC(F)(F)F)C(C(F)(F)F)O
FC(F)(F)Oc1cccc(c1)C3(CN(C[C@@H](O)C(F)(F)F)c2ccccc23)Cc4cccc(OC(F)(F)F)c4
///////////see..........http://newdrugapprovals.org/2016/01/06/mercks-novel-indoline-cholesterol-ester-transfer-protein-inhibitors-cetp/
Indoline 7 as in ACS MEDCHEM LETTERS, DOI: 10.1021/acsmedchemlett.5b00404
and
eg 10 as in WO2015054088
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
1H-Indole-1-ethanol, 2,3-dihydro-3-[3-(trifluoromethoxy)phenyl]-3-[[3- (trifluoromethoxy)phenyl]methyl]-α-(trifluoromethyl)-, (αR)-
cas 1699732-96-1 R ISOMER
MF C26 H20 F9 N O3, MW 565.43
Merck Sharp & Dohme Corp. INNOVATOR
Using the collective body of known
(CETP) inhibitors as inspiration for design, a structurally novel series
of tetrahydroquinoxaline CETP inhibitors were discovered. An exemplar
from this series, compound 5, displayed potent in vitro CETP
inhibition and was efficacious in a transgenic cynomologus-CETP mouse
HDL PD (pharmacodynamic) assay. However, an undesirable metabolic
profile and chemical instability hampered further development of the
series. A three-dimensional structure of tetrahydroquinoxaline inhibitor
6 was proposed from 1H NMR structural studies, and
this model was then used in silico for the design of a new class of
compounds based upon an indoline scaffold. This work resulted in the
discovery of compound 7, which displayed potent in vitro CETP inhibition, a favorable PK–PD profile relative to tetrahydroquinoxaline 5, and dose-dependent efficacy in the transgenic cynomologus-CETP mouse HDL PD assay.
chemical compounds that inhibit cholesterol
ester transfer protein (CETP) and are expected to have utility in
raising HDL-C, lowering LDL-C, and in the treatment and prevention of
atherosclerosis.see………….http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00404
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.5b00404/suppl_file/ml5b00404_si_001.pdf
Discovery of Novel Indoline Cholesterol Ester Transfer Protein Inhibitors (CETP) through a Structure-Guided Approach
†Department of Medicinal Chemistry and ‡Department of Structural Chemistry, Merck Research Laboratories, Merck & Co, Inc., P.O. Box 2000, Rahway, New Jersey 07065, United States
§Department of Pharmacology, ∥Department of Drug Metabolism and Pharmacokinetics, and ⊥Department of Biology, Merck Research Laboratories, Merck & Co, Inc., P.O. Box 2000, Kenilworth, New Jersey 07033, United States
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.5b00404
Publication Date (Web): January 4, 2016
Copyright © 2016 American Chemical Society
*E-mail: jonathan_wilson@merck.com.
PATENT
Atherosclerosis and its clinical consequences, including coronary heart disease
(CHD), stroke and peripheral vascular disease, represent a truly enormous burden to the health care systems of the industrialized world. In the United States alone, approximately 13 million patients have been diagnosed with CHD, and greater than one half million deaths are attributed to CHD each year. Further, this toll is expected to grow over the next quarter century as an epidemic in obesity and diabetes continues to grow.
It has long been recognized that in mammals, variations in circulating lipoprotein profiles correlate with the risk of atherosclerosis and CHD. The clinical success of HMG-CoA reductase inhibitors, especially the statins, in reducing coronary events is based on the reduction of circulating low density lipoprotein cholesterol (LDL-C), levels of which correlate directly with an increased risk for atherosclerosis. More recently, epidemiologic studies have
demonstrated an inverse relationship between high density lipoprotein cholesterol (HDL-C) levels and atherosclerosis, leading to the conclusion that low serum HDL-C levels are associated with an increased risk for CHD.
Metabolic control of lipoprotein levels is a complex and dynamic process involving many factors. One important metabolic control in man is the cholesteryl ester transfer protein (CETP), a plasma glycoprotein that catalyzes the movement of cholesteryl esters from HDL to the apoB containing lipoproteins, especially VLDL (see Hesler, C.B., et. al. (1987) Purification and characterization of human plasma cholesteryl ester transfer protein. J. Biol. Chem. 262(5), 2275-2282)). Under physiological conditions, the net reaction is a heteroexchange in which CETP carries triglyceride to HDL from the apoB lipoprotein and transports cholesterol ester from HDL to the apoB lipoprotein.
In humans, CETP plays a role in reverse cholesterol transport, the process whereby cholesterol is returned to the liver from peripheral tissues. Intriguingly, many animals do not possess CETP, including animals that have high HDL levels and are known to be resistant to coronary heart disease, such as rodents (see Guyard-Dangremont, V., et. al, (1998)
Phospholipid and cholesteryl ester transfer activities in plasma from 14 vertebrate species. Relation to atherogenesis susceptibility, Comp. Biochem. Physiol. B Biochem. Mol. Biol. 120(3), 517-525). Numerous epidemiologic studies correlating the effects of natural variation in CETP activity with respect to coronary heart disease risk have been performed, including studies on a small number of known human null mutations (see Hirano, K.-L, Yamashita, S. and Matsuzawa, Y. (2000) Pros and cons of inhibiting cholesteryl ester transfer protein, Curr. Opin. Lipidol. 11(6), 589-596). These studies have clearly demonstrated an inverse correlation between plasma HDL-C concentration and CETP activity (see Inazu, A., et. al. (2000) Cholesteryl ester transfer protein and atherosclerosis, Curr. Opin. Lipidol. 11(4), 389-396), leading to the hypothesis that pharmacologic inhibition of CETP lipid transfer activity may be beneficial to humans by increasing levels of HDL-C while lowering LDL-C.
Despite the significant therapeutic advance that statins such as simvastatin and atorvastatin represent, statins only achieve a risk reduction of approximately one-third in the treatment and prevention of atherosclerosis and ensuing atherosclerotic disease events.
Currently, few pharmacologic therapies are available that favorably raise circulating levels of HDL-C. Certain statins and some fibrates offer modest HDL-C gains. Niacin provides an effective therapy for raising HDL-C but suffers from patient compliance issues, due in part to side effects such as flushing. Drugs that inhibit CETP (CETP inhibitors) have been under development with the expectation that they will effectively raise HDL cholesterol levels and also reduce the incidence of atherosclerosis in patients. Torcetrapib was the first drug that was tested in a long-term outcomes clinical trial. The clinical trial of torcetrapib was terminated early due to a higher incidence of mortality in patients to whom torcetrapib and atorvastatin were administered concomitantly compared with patients who were treated with atorvastatin alone. The cause of the increased mortality is not completely understood, but it is not believed to be associated with the CETP inhibiting effects of the drug.
Two other drug candidates, dalcetrapib and anacetrapib, are currently being tested in Phase III clinical trials, including large scale outcomes trials. Data from the recently completed DEFINE Phase III trial of anacetrapib are promising. Patients who were being treated with anacetrapib along with baseline statin therapy showed an increase of HDL-C of 138% and a decrease of LDL-C of 40%> compared with patients who were treated with just a statin. See: N. Engl. J. Med. 2010: 363: 2406-15. The data in the DEFINE trial were sufficient to indicate that an increase in mortality for patients treated with anacetrapib is unlikely. Additional drug candidates are still being sought that may have properties that are advantageous compared with the CETP inhibitors that have so far been studied or are currently being studied. Such properties may include, for example, higher potency, reduced off-target activity, better pharmacodynamics, higher bioavailability, or a reduced food effect compared with many of the highly lipophilic compounds that have so far been studied. “Food effect” refers to the variability in exposure to the active drug that occurs depending on when the patient had last eaten, whether or not the drug is administered with food, and the fat content of the food.
(CHD), stroke and peripheral vascular disease, represent a truly enormous burden to the health care systems of the industrialized world. In the United States alone, approximately 13 million patients have been diagnosed with CHD, and greater than one half million deaths are attributed to CHD each year. Further, this toll is expected to grow over the next quarter century as an epidemic in obesity and diabetes continues to grow.
It has long been recognized that in mammals, variations in circulating lipoprotein profiles correlate with the risk of atherosclerosis and CHD. The clinical success of HMG-CoA reductase inhibitors, especially the statins, in reducing coronary events is based on the reduction of circulating low density lipoprotein cholesterol (LDL-C), levels of which correlate directly with an increased risk for atherosclerosis. More recently, epidemiologic studies have
demonstrated an inverse relationship between high density lipoprotein cholesterol (HDL-C) levels and atherosclerosis, leading to the conclusion that low serum HDL-C levels are associated with an increased risk for CHD.
Metabolic control of lipoprotein levels is a complex and dynamic process involving many factors. One important metabolic control in man is the cholesteryl ester transfer protein (CETP), a plasma glycoprotein that catalyzes the movement of cholesteryl esters from HDL to the apoB containing lipoproteins, especially VLDL (see Hesler, C.B., et. al. (1987) Purification and characterization of human plasma cholesteryl ester transfer protein. J. Biol. Chem. 262(5), 2275-2282)). Under physiological conditions, the net reaction is a heteroexchange in which CETP carries triglyceride to HDL from the apoB lipoprotein and transports cholesterol ester from HDL to the apoB lipoprotein.
In humans, CETP plays a role in reverse cholesterol transport, the process whereby cholesterol is returned to the liver from peripheral tissues. Intriguingly, many animals do not possess CETP, including animals that have high HDL levels and are known to be resistant to coronary heart disease, such as rodents (see Guyard-Dangremont, V., et. al, (1998)
Phospholipid and cholesteryl ester transfer activities in plasma from 14 vertebrate species. Relation to atherogenesis susceptibility, Comp. Biochem. Physiol. B Biochem. Mol. Biol. 120(3), 517-525). Numerous epidemiologic studies correlating the effects of natural variation in CETP activity with respect to coronary heart disease risk have been performed, including studies on a small number of known human null mutations (see Hirano, K.-L, Yamashita, S. and Matsuzawa, Y. (2000) Pros and cons of inhibiting cholesteryl ester transfer protein, Curr. Opin. Lipidol. 11(6), 589-596). These studies have clearly demonstrated an inverse correlation between plasma HDL-C concentration and CETP activity (see Inazu, A., et. al. (2000) Cholesteryl ester transfer protein and atherosclerosis, Curr. Opin. Lipidol. 11(4), 389-396), leading to the hypothesis that pharmacologic inhibition of CETP lipid transfer activity may be beneficial to humans by increasing levels of HDL-C while lowering LDL-C.
Despite the significant therapeutic advance that statins such as simvastatin and atorvastatin represent, statins only achieve a risk reduction of approximately one-third in the treatment and prevention of atherosclerosis and ensuing atherosclerotic disease events.
Currently, few pharmacologic therapies are available that favorably raise circulating levels of HDL-C. Certain statins and some fibrates offer modest HDL-C gains. Niacin provides an effective therapy for raising HDL-C but suffers from patient compliance issues, due in part to side effects such as flushing. Drugs that inhibit CETP (CETP inhibitors) have been under development with the expectation that they will effectively raise HDL cholesterol levels and also reduce the incidence of atherosclerosis in patients. Torcetrapib was the first drug that was tested in a long-term outcomes clinical trial. The clinical trial of torcetrapib was terminated early due to a higher incidence of mortality in patients to whom torcetrapib and atorvastatin were administered concomitantly compared with patients who were treated with atorvastatin alone. The cause of the increased mortality is not completely understood, but it is not believed to be associated with the CETP inhibiting effects of the drug.
Two other drug candidates, dalcetrapib and anacetrapib, are currently being tested in Phase III clinical trials, including large scale outcomes trials. Data from the recently completed DEFINE Phase III trial of anacetrapib are promising. Patients who were being treated with anacetrapib along with baseline statin therapy showed an increase of HDL-C of 138% and a decrease of LDL-C of 40%> compared with patients who were treated with just a statin. See: N. Engl. J. Med. 2010: 363: 2406-15. The data in the DEFINE trial were sufficient to indicate that an increase in mortality for patients treated with anacetrapib is unlikely. Additional drug candidates are still being sought that may have properties that are advantageous compared with the CETP inhibitors that have so far been studied or are currently being studied. Such properties may include, for example, higher potency, reduced off-target activity, better pharmacodynamics, higher bioavailability, or a reduced food effect compared with many of the highly lipophilic compounds that have so far been studied. “Food effect” refers to the variability in exposure to the active drug that occurs depending on when the patient had last eaten, whether or not the drug is administered with food, and the fat content of the food.
(R)- 1,1, 1 -trifluoro-3-((R)-4-(3-trifluoromethoxy)benzyl)-2-(3-(l, 1 ,2,2,-tetrafluoroethoxy)phenyl)-3,4- dihydroquinoxalin- 1 (2H)-yl)propan-2-ol
SPA: 15 nM
Example 18 was prepared from 2-bromo-l-(3-(l , 1 ,2,2,-tetrafluoroethoxy)phenyl)ethanone in three steps, using the reactions detailed in Schemes A6, A2 and Al . Spectral data are as follows: 1H NMR (400 MHz, CDC13) £2.70 (bd, J=4.1 Hz, IH), 3.24 (dd, J=l 1.3, 3.4 Hz, IH), 3.34 (dd, J=15.5, 9.7 Hz, IH), 3.58 (dd, J=l 1.3, 3.3 Hz, IH), 3.86 (d, J=15.4 Hz, IH), 4.20 (d, J=15.7 Hz, IH), 4.40 (d, J=15.8 Hz, IH), 4.46 (m, IH), 4.927 (t, J=3.3 Hz, IH), 5.90 (tt, J=53.1 , 2.7 Hz, IH), 6.59 (d, J= 7.9 Hz, IH), 6.72 (m, 2H), 6.84 (m, 2H), 6.92 (d, J=7.6 Hz, IH), 7.20 (m, 2H), 7.35 (t, J=7.9 Hz, IH), MS m/z = 613.03.
Scheme A12
Methyl 3 – { 1 – [(R)-3 ,3 ,3 -trifluoro-2-hy droxypropyl] -4- [3 -(trifluoromethoxy) benzyl]-l,2,3,4-tetrahydroquinoxalin-2-yl}benzoate (700 mg, 1.262 mmol) is made as described in Example 16 but with one stereochemical center unresolved. The compound was dissolved in MeOH (12.6mL), lithium hydroxide monohydrate (530 mg, 12.62 mmol) was added, and the reaction mixture was heated to 60°C for 4 hours. The crude mixture was dissolved in saturated ammonium chloride solution and extracted into EtOAc, the organic phase was dried with anhydrous magnesium sulfate, filtered, concentrated, and purified on a silica gel column with a 0-100% Hex/EtOAc gradient. The major peak was concentrated to afford 3-{l-[(R)-3,3,3-trifluoro-2-hydroxypropyl]-4-[3-(trifluoromethoxy)benzyl]-l,2,3,4-tetra-hydroquinoxalin-2-yl} benzoic acid. MS m/z = 541.09.
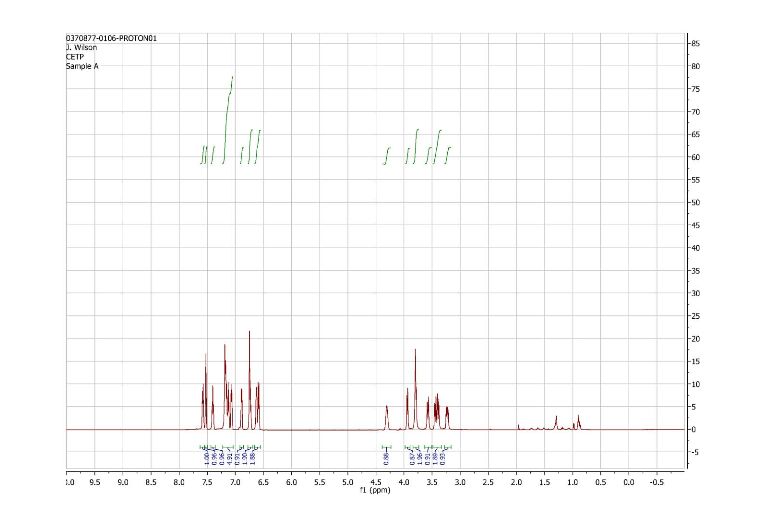
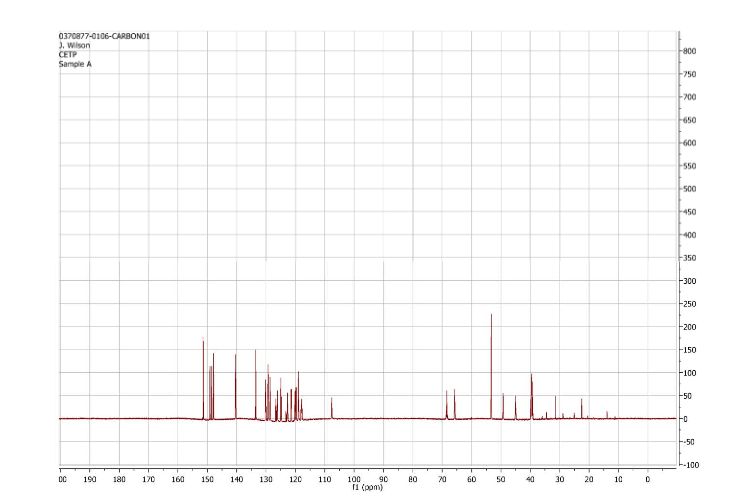
1H and 13C NMR spectra for compound 7
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
Patent
WO2015054088
http://google.com/patents/WO2015054088A1?cl=en
Scheme Al
Scheme A2
Scheme A3
R = Ar, NR2l C02R, CN, S02Me
es
es
SEE EXAMPLE ………SIMILAR BUT NOT SAME
Example 1. (2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. This material was prepared according to Scheme Al, as described below.
3-(3-(trifluoromethyl)phenyl)indolin-2-one. Oxindole (1.598 g, 12 mmol), 3-bromo-a,a,a-trifluoromethyltoluene (2.009 ml, 14.40 mmol), potassium carbonate (3.32 g, 24.00 mmol), Pd2dba3 (0.220 g, 0.240 mmol), and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (0.458 g, 0.960 mmol) were combined in THF (12 ml) and the mixture was degassed with nitrogen. The solution was then heated to 80 °C for 18h. The mixture was cooled to room temperature, filtered through silica eluting with ethyl acetate, and concentrated. The material was then purified by silica gel chromatography (Biotage lOOg SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethyl)phenyl)indolin-2-one as a white solid.
1H NMR (500 MHz) δ 8.58 (s, 1H), 7.61 (d, J=7 Hz, 1H), 7.53-7.45 (m, 3H), 7.33-7.29 (m, 1H), 7.16 (d, J=7 Hz, 1H), 7.10 (m, 1H), 7.01-6.90 (m, 1H), 4.73 (s, 1H).
3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin-2-one . 3 -Trifluoromethoxy-benzylbromide (0.204 ml, 1.255 mmol) was added to a mixture of 3-(3-(trifluoromethyl)-phenyl)indolin-2-one (290 mg, 1.046 mmol) and potassium carbonate (289 mg, 2.092 mmol) (sodium carbonate may be used in place of potassium carbonate) in DMA (2.5 ml). The mixture was stirred at r.t. for 16h. The reaction was diluted with ethyl acetate and washed with water (3×5 mL). The organic layer was dried with Na2S04, filtered, and concentrated. The products were then purified by silica gel chromatography (Biotage 50g SNAP cartridge; 0-40%> ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-2-one .
1H NMR (500 MHz) δ 7.79 (s, 1H), 7.73 (d, J=7 Hz, 1H), 7.62-7.60 (m, 2H), 7.51 (t, J=7 Hz, 1H), 7.26- 7.22 (m, 2H), 7.14 (t, J=7.0 Hz, 1H), 7.11 (m, 1H), 6.97 (m, 1H), 6.92 (m, 1H), 6.78 (m, 1H), 6.73 (s, 1H), 3.77 (d, J=13 Hz, 1H), 3.49 (d, J=13 Hz, 1H).
LCMS m/z = 451.8 (M+H)
3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. Borane tetrahydrofuran complex (1.673 ml, 1.673 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indolin-2-one (302 mg, 0.669 mmol) in THF (1.5 ml). The mixture was heated to 70 °C for 20h. The reaction was cooled to room temperature and quenched with saturated NH4C1 solution, and this mixture was stirred vigorously for 20 minutes. The product was extracted with ethyl acetate. The extracts were dried over Na2S04, filtered, and concentrated. The product was purified by silica gel chromatography (Biotage 25g SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. This material may also be used without purification in the final step of the sequence, epoxide opening.
1H NMR (500 MHz) δ 7.66 (s, IH), 7.59 (d, J=7 Hz, IH), 7.53 (d, J=7 Hz, IH), 7.45 (t, J=8 Hz, IH), 7.18-7.13 (m, 2H), 7.04 (d, J=8 Hz, IH), 6.98 (d, J=7 Hz, IH), 6.81 (t, J=7.5 Hz, IH), 6.71 (m, 2H), 6.60 (s, IH), 3.83 (m, IH), 3.75-3.73 (m, 2H), 3.46 (d, J=13 Hz, IH), 3.41 (d, J=13 Hz, IH).
= 437.9 (M+H)
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. (S)-2-(trifluoromethyl)oxirane (81 μΐ, 0.933 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline (136 mg, 0.311 mmol) in l,l,l,3,3,3-hexafluoro-2-propanol (412 μΐ, 3.91 mmol). The reaction was stirred at room temperature overnight. The solvent was removed and the product was purified by silica gel chromatography (Biotage 25 g SNAP cartridge; 0-25% ethyl acetate in hexanes) to provide (2R)- 1 ,1,1 -trifluoro-3 -(3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin- 1 -yl)propan-2-ol.
1H NMR (500 MHz) (mixture of diastereomers) δ 7.72 (s, 0.5 H), 7.69 (s, 0.5 H), 7.65 (d, J=6.5 Hz, 0.5 H), 7.61 (d, J=7.5 Hz, 0.5 H), 7.56 (s, 1H), 7.50 (m, 1H), 7.25-7.17 (m, 2H), 7.07 (broad s, 2H), 6.91-6.89 (m, 1H), 6.79-6.75 (m, 1H), 6.53 (m, 2H), 4.00 (broad s, 1H), 3.83 (d, J= 9 Hz, 0.5H), 3.77 (d, J=9 Hz, 0.5H), 3.59-3.55 (m, 1H), 3.45-3.43 (m, 1H), 3.39-3.29 (m, 2H), 3.21-3.15 (m, 1H), 2.32 (m, 0.5H), 2.15 (m, 0.5H).
LCMS m/z = 549.8 (M+H)
Examples 1-25, in the table below, were prepared according to Scheme Al in a
SEE EG 10…….(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
ABOUT AUTHOR
Jonathan Wilson
Associate Principal Scientist at Merck
Experience
Associate Principal Scientist
Merck
Senior scientist
Merck

Postdoctoral researcher
Princeton University
Associate Medicinal Chemist
Merck
Education


c21ccccc1N(C[C@@]2(c3cccc(c3)OC(F)(F)F)Cc4cc(ccc4)OC(F)(F)F)C(C(F)(F)F)O
FC(F)(F)Oc1cccc(c1)C3(CN(C[C@@H](O)C(F)(F)F)c2ccccc23)Cc4cccc(OC(F)(F)F)c4
///////////see..........http://newdrugapprovals.org/2016/01/06/mercks-novel-indoline-cholesterol-ester-transfer-protein-inhibitors-cetp/