
TEMSIROLIMUS
Proline CCI-779
Torisel, NCGC00167518-01
LAUNCHED 2007
PFIZER
- CCI 779
- CCI-779
- HSDB 7931
- Temsirolimus
- Torisel
- UNII-624KN6GM2T
- WAY-CCI 779
Inhibits mTOR protein
For the treatment of renal cell carcinoma (RCC). Also investigated for use/treatment in breast cancer, lymphoma (unspecified), rheumatoid arthritis, and multiple myeloma.
An ester analog of rapamycin. Temsirolimus binds to and inhibits the mammalian target of rapamycin (mTOR), resulting in decreased expression of mRNAs necessary for cell cycle progression and arresting cells in the G1 phase of the cell cycle. mTOR is a serine/threonine kinase which plays a role in the PI3K/AKT pathway that is upregulated in some tumors
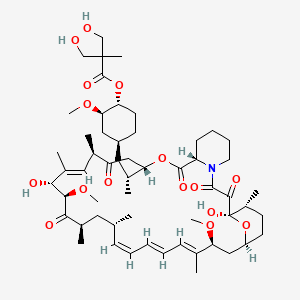
(1R,2R,4S)-4-{(2R)-2-[(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-1,5,11,28,29-pentaoxo-1,4,5,6,9,10,11,12,13,14,21,22,23,24,25,26,27,28,29,31,32,33,34,34a-tetracosahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontin-3-yl]propyl}-2-methoxycyclohexyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate
cas 162635-04-3
Temsirolimus is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by Wyeth Pharmaceuticals and approved by the FDA in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
Molecular Formula: C56H87NO16
Molecular Weight: 1030.28708
Temsirolimus (CCI-779) is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by WyethPharmaceuticals and approved by the U.S. Food and Drug Administration (FDA) in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
 TEMSIROLIMUS
TEMSIROLIMUS
Temsirolimus is a specific inhibitor of mTOR and interferes with the synthesis of proteins that regulate proliferation, growth, and survival of tumor cells. Treatment with temsirolimus leads to cell cycle arrest in the G1 phase, and also inhibits tumor angiogenesis by reducing synthesis of VEGF.
The product had been under development by Wyeth Pharmaceutical for the treatment of pancreas cancer and metastatic breast cancer, multiple sclerosis (MS) and rheumatoid arthritis (RA); however, no recent development for these indications has been reported. Pfizer had been developing the compound for the treatment of sarcoma.
Temsirolimus holds orphan drug designation in both the U.S. and the E.U. for the treatment of renal cell carcinoma. Orphan drug designation was received in the U.S. in 2006 for the treatment of mantle-cell lymphoma.
mTOR (mammalian target of rapamycin) is a kinase enzyme inside the cell that collects and interprets the numerous and varied growth and survival signals received by tumor cells. When the kinase activity of mTOR is activated, its downstream effectors, the synthesis of cell cycle proteins such as cyclin D and hypoxia-inducible factor-1a (HIF-1a) are increased. HIF-1a then stimulates VEGF. Whether or not mTOR kinase is activated, determines whether the tumor cell produces key proteins needed for proliferation, growth, survival, and angiogenesis.
mTOR is activated in tumor cells by various mechanisms including growth factor surface receptor tyrosine kinases, oncogenes, and loss of tumor suppressor genes. These activating factors are known to be important for malignant transformation and progression.mTOR is particularly important in the biology of renal cancer (RCC) owing to its function in regulating HIF-1a levels. Mutation or loss of the von Hippel Lindau tumor-suppressor gene is common in RCC and is manifested by reduced degradation of HIF-1a. In RCC tumors, activated mTOR further exacerbates accumulation of HIF-1a by increasing synthesis of this transcription factor and its angiogenic target gene products.
Rapamycin 42-ester with 3-hydroxy-2-(hydroxymethyl)-2-methylpropionic acid (CCl-779) is an ester of rapamycin which has demonstrated significant inhibitory effects on tumor growth in both in vitro and in vivo models.
CCl-779 may delay the time to progression of tumors or time to tumor recurrence which is more typical of cytostatic rather than cytotoxic agents. CCl-779 is considered to have a mechanism of action that is similar to that of sirolimus. CCl-779 binds to and forms a complex with the cytoplasmic protein FKBP, which inhibits an enzyme, mTOR (mammalian target of rapamycin, also known as FKBP12-rapamycin associated protein [FRAP]). Inhibition of mTOR's kinase activity inhibits a variety of signal transduction pathways, including cytokine-stimulated cell proliferation, translation of mRNAs for several key proteins that regulate the G1 phase of the cell cycle, and IL-2-induced transcription, leading to inhibition of progression of the cell cycle from G1 to S. The mechanism of action of CCl-779 that results in the G1-S phase block is novel for an anticancer drug.
The preparation and use of hydroxyesters of rapamycin, including CCl-779, are disclosed in U.S. Pat. No. 5,362,718. A regiospecific synthesis of CCl-779 is described in U.S. Pat. No. 6,277,983.
CCl-779 can be synthesized by the non-regioselective acylation of rapamycin, as described in U.S. Pat. No. 5,362,718. The synthesis, however, is complicated by mixtures of the desired 42-ester, with 31-esterified rapamycin, as well as 31, 42-diesterified rapamycin and unreacted rapamycin.
CCl-779 can also be prepared by the acylation of the 31-silyl ether of rapamycin with a ketal of bis-(hydroxymethyl)propionic acid, followed by removal of the 31-silyl ether and ketal protecting group from the bis-(hydroxymethyl) propionic acid, as described in U.S. Pat. No. 6,277,983. However, the crude 42-monoester produced from this regioselective synthesis requires further purification by column chromatography to remove residual amounts of diester by-products and unreacted rapamycin starting material.
Temsirolimus (CCI-779), an mTOR kinase Inhibitor of formula (I) is an antineoplastic agent indicated for the treatment of advanced renal cell carcinoma.Temsirolimus is a Rapamycin 42 ester with [3-hydroxy-2-(hydroxymethyl)-2-methylpropanoic acid and was first disclosed by Skotnicki et al in US Patent No. 5,362,718.

Several processes for the preparation of Temsirolimus have been reported in the literature such as those described in US 5,362,718; US 6,277,983 and US 7, 153,957.
US Patent No 5,362,718 discloses a process for the preparation of different rapamycin 42 esters including Temsirolimus as per the scheme given below (Scheme-I).

Scheme-I: Synthesis of Temsirolimus as disclosed in US Patent No. 5,362,718
The process is non-regioselective and hence results in 31-estehfied rapamycin, 31 , 42 diesterified rapamycin and unreacted rapamycin along with the desired rapamycin-42 ester.
US Patent No. 6,277,983 reports a process for the preparation of Temsirolimus by using 31 , 42 bis silyl intermediates as per the scheme shown below (Scheme-ll).

Scheme-ll: Synthesis of Temsirolimus as disclosed in US Patent No. 6,277,983 US Patent No. 7, 153,957 reports a process for the preparation of Temsirolimusby using boronate intermediate as per the scheme shown below (Scheme-Ill).

Scheme-Ill: Synthesis of Temsirolimus as disclosed in US Patent No. 7, 153,957
 Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS "Amano" (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.
Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS "Amano" (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.
........................................................
SYNTHESIS
Example 11
Rapamycin 42-ester with 2.2-bis-(hydroxymethyl)propionic acid
A solution of the product of Example 10 (2.8 g, 2.65 mmol) in 50 mL THF and
25 mL IN HCl was stirred at room temperature for 4 h. The mixture was diluted with water and extracted three times with EtOAc. The combined organic phases were washed with saturated NaHCO3 solution, saturated NaCl solution, dried over MgSO4, filtered and evaporated to a yellow oily solid. Purification by flash chromatography (3X with EtOAc) afforded the title compound (1.6 g, 59 %).
(-)FAB-MS mlz 1029.6 (M-), 590.4 (southern fragment), 437.3 (northern fragment). !H NMR (400 MHz, d-6 DMSO) δ 4.5 (m, 1 H, C(42)H), 3.45 (s, 4 H), 1.04 (s, 3 H).
*3C NMR (100.6 MHz, d-6 DMSO) δ 174.2, 63.7, 63.6, 49.9, 16.8.
Example 10 Rapamycin 42-ester with 2.2.5-trimethyl.1.3_dioxane-5-carboxyric acid
To a solution of the 2,2-bis(hydroxymethyl)propionic acid isopropylidene ketal (1.041 g, 5.98 mmol) (prepared according to the procedure of Bruice, J. Am. Chem. Soc. 89: 3568 (1967)) and triethylamine (0.83 mL, 5.98 mmol) in 20 mL anhydrous THF at 0 °C under nitrogen was added 2, 4, 6-trichlorobenzoyl chloride (0.93 mL, 5.98 mmol) and the resultant white suspension was stirred 5 h at room temperature. The precipitate was removed by vacuum filtration, rinsing the flask and filter cake with an additional 10 mL dry THF. The filtrate was concentrated by rotary evaporation to a white solid. The residue was dissolved in 20 mL dry benzene, then rapamycin (5.47 g, 5.98 mmol) and DMAP (0.731 g, 5.98 mmol) were added. After stirring overnight at room temperature, the mixture was diluted with EtOAc, washed with H2O and saturated NaCl (aq), dried over MgSO4, filtered and evaporated to a yellow oil. Flash chromatography (5X with 60% EtOAc-hexane) afforded the title compound (2.2 g, 34 %) as a white solid.
(-)FAB-MS mlz 1069.5 (M-), 590.3 (southern fragment), 477.2 (northern fragment). -■H NMR (400 MHz, d-6 DMSO) δ 4.57 (m, 1 H, C(42)H), 4.02 (d, 2 H), 3.60 (d, 2 H), 1.34 (s, 3 H), 1.24 (s, 3 H), 1.06 (s, 3 H). 1 C NMR (100.6 MHz, d-6 DMSO) δ 173.2, 99.0, 65.0, 22.2, 18.1.
..................................................
SYNTHESIS
This scheme



Preparation of 5-Methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [A]
To a suspension of 2,2-bis(hydroxymethyl)propionic acid (131 g, 0.98 mole) in tetrahydrofuran (500 ml) was added a solution of phenylboronic acid (122 g, 1.0 mole) in tetrahydrofuran (500 ml). The mixture was stirred for 3 h and toluene (1.0 L) was added. Water was removed by azeotropic distillation with toluene. Heptanes (500 ml) was added to the precipitated product, heated to reflux and cooled. The mixture was filtered and washed with heptanes (2×300 ml). The solids were dried under vacuum at 70–75° C. until constant weight to give 94% yield. 1H NMR: δ (DMSO-d6) 7.65 (d, 2H, Ar), 7.40 (m, 3H, Ar), 4.35 (d, 2H, CH2), 3.92 (d, 2H, CH2), 1.17 (s, 3H, CH3)
Preparation of Rapamycin 42-ester with 5-methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [B]
As described in U.S. Pat. No. 6,277,983 (2001) a 3 L flask was charged with rapamycin (100 g, 0.104 mole) and dissolved in ethyl acetate (1.50 L). The solution was cooled to 5–10° C. Imidazole (30 g, 0.44 moles, 4.23 eq.) was added and dissolved. Under nitrogen protection, trimethylsilyl chloride (44 g, 0.405 mole, 4.0 eq.) was added over 30–40 min while maintaining the temperature at 0–5° C. during the addition. The mixture was held for a minimum of 0.5 h. The reaction was monitored by TLC (30:70 acetone:heptane eluent). The reaction was complete when all of the rapamycin was consumed.
Two to three drops of the reaction mixture were removed and retained as a 31,42-bis(trimethylsilyl) rapamycin reference standard. 0.5 N Sulfuric acid (300 mL) was added to the 3 L flask over 0.5 h maintaining the temperature 0–5° C. The mixture was stirred vigorously and held for 5 h. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone:heptane eluent). The reaction was complete when essentially no 31,42-bis-(trimethylsilyl) rapamycin was present. The layers were separated and the lower aqueous layer was back extracted with ethyl acetate (500 mL). The combined organic layers were washed with saturated brine (500 mL) and saturated sodium bicarbonate (2×200 mL) until pH 8 was obtained. The organic layer was washed with water (2×500 mL) and brine (500 ml) until pH 6 to 7 was obtained. The solution was dried over magnesium sulfate (100 g) for 30 min, filtered into a 2 L flask and concentrated to a volume of 135 ml. Ethyl acetate (500 ml) was added and concentrated to a volume of 135 ml. The water chase was repeated once more with ethyl acetate (500 ml). Methylene chloride (300 ml) was added and the solution held until needed in the next step.
A 3 L flask equipped with mechanical stirrer was charged with compound [A] (75 g, 0.341 mole) in methylene chloride (400 mL). Diisopropylethylamine (66.1 g, 0.51 mole) was added dropwise over 20 mins and rinsed with methylene chloride (25 mL). 2,4,6-Trichlorobenzoyl chloride (80 g, 0.328 mole) was added and rinsed with methylene chloride (25 mL). The mixture was held at 0–5° C. for 4 h, and cooled to −10±5° C.
The solution of 31-trimethylsilyl rapamycin was added to the 3 L flask containing the mixed anhydride, and rinsed with methylene chloride (25 mL). A solution of dimethylamino pyridine (48.5 g, 0.397 mole) in methylene chloride (150 mL) was prepared, added over 1.5 h, maintaining the temperature <−8° C., and rinsed with methylene chloride (25 mL). The mixture was held for 12 h at −11 to −5° C. The reaction mixture was quenched with 1 N sulfuric acid (600 ml) keeping the temperature <10° C. The mixture was stirred and held for 30 mins. The pH of the upper aqueous layer was ≦2. The layers were separated, and the lower organic layers washed with brine (450 ml), saturated sodium bicarbonate (500 mL) until pH ≧8. The organic layer was washed with water (450 ml) until pH 6–7 was obtained. The solution was concentrated, acetone (250 ml) added and concentrated. This was repeated with another portion of acetone (250 ml) and concentrated.
The solution was diluted with acetone. 0.5 N Sulfuric acid (500 ml) was added dropwise over 30 mins keeping the pot temperature 0–5° C. The mixture was held for a minimum of 5 h, during which time, the product precipitated out of solution. Aqueous sodium bicarbonate (30 g in 375 ml water) was added dropwise over 30 minutes keeping the pot temperature 0 to 5° C.; the mixture was held for a minimum of 30 minutes. Acetic acid (25 ml) was added until pH was 5–6 keeping the pot temperature <10° C. The mixture was warmed to room temperature and held for 16 h. The solid product was filtered and washed with water (2×100 ml) followed by 1:1 acetone:water (2×100 ml). The cake was purified in acetone (375 ml) to give 65 g (58% overall from rapamycin) of product [B]. LC/MS: using an electrospray interface in the positive ion mode afforded the molecular ion [M+Na]=1138.5 atomic mass units (amu).
Preparation of Rapamycin 42-ester with 2,2-bis(hydroxymethyl)-propionic acid, [C]
Compound [B] (200 g, 0.179 mole), was dissolved in tetrahydrofuran (600 ml), 2-methyl-2,4-pentanediol (42.3 g, 0.358 mole, 2.0 eq.) was added and the mixture stirred for a minimum of 3 h. The reaction mixture was concentrated to a foam. Diethyl ether (1.0 L) was added and the mixture stirred for 2 h. Heptanes (1.0 L) was added dropwise over 1 h and the mixture stirred for 2 h. The mixture was filtered and the solid product washed with heptanes (500 ml). The solids were re-dissolved in acetone (400 ml), re-treated with 2-methyl-2,4-pentanediol (21.1 g, 0.179 mole, 1 eq.) in acetone (200 ml), clarified through a 0.2 micron cartridge filter, and rinsed with acetone (200 ml). The solution was concentrated to a foam, diethyl ether (1.0 L), pre-filtered through a 0.2 micron cartridge filter, was added and the mixture stirred for 2 h. The mixture was co-precipitated by adding pre-filtered heptanes (1.0 L). The precipitated solids were filtered and washed with ether:heptane (2×500 ml). The solids were dried (55 to 60° C., 10 mm Hg, minimum 24 h) to give 159 g (86%) of product [C]. LC/MS: using APCl in the positive ion mode afforded the molecular ion [M+NH4]=1047.0 amu. The 1H NMR of the product (CCl-779) was identical to the product described in example 11 of U.S. Pat. No. 5,362,718 (1994).
.......................................
Synthesis
Example 1 - Synthesis of Proline CCI-779

This example describes a method for the synthesis of the proline analog of CCI- 779, which is illustrated in the scheme provided above.
A.
Preparation of 31, 42-Bis (trimethylsilyl) proline rapamycin (Compound B)
A 3 -neck 50 mL flask was charged with proline rapamycin (compound A in the scheme) (1.47 g, 1.63 mmol), imidazole (0.45 g, 6.6 mmol, 4 eq.) and ethyl acetate (22.5 mL). The magnetically stirred mixture became cloudy. The mixture was cooled to 0-5°C. Under nitrogen protection, trimethylsilyl chloride (0.62 g, 5.7 mmol, 3.5 eq.) was added over 0.5 h via syringe while maintaining the temperature at 0-5°C during the addition. The syringe was rinsed with 2.5 ml ethyl acetate and the mixture held for 0.75 hours (0.75 h), whereupon a white precipitate was formed. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone :heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The mixture was shaken and allowed to settle. The upper organic layer was spotted against the starting material (proline rapamycin). The reaction was complete when no more starting material was present.
B.
Preparation of 31 -trimethylsilyl proline rapamycin, Compound E
When the above reaction was complete, 2-3 drops of the reaction mixture was removed and retained for the following step as the 31,42-bis(trimethylsilyl) proline rapamycin reference standard. To the 50 ml flask was added 0.5 N sulfuric acid (4.5 mL) over 0.5 h maintaining the temperature at 0-5 °C. The mixture became less cloudy. The mixture was held for 2.5 h and was monitored by thin layer chromatography (TLC, 30:70 acetone:heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The reaction aliquot was shaken and allowed to settle. The upper organic layer was spotted against the 31 ,42-bis(trimethylsilyl) proline rapamycin reference. The reaction was complete when essentially no 31,42-bis(trimethylsilyl) proline rapamycin was present. Ethyl acetate (5 mL) was added and the layers separated. The lower aqueous layer is extracted with ethyl acetate (7.5 mL). The combined organic layers were washed with brine (7.5 mL), by washing with saturated sodium bicarbonate (6 mL) followed by washing water (3 x 7.5 mL), in that order. The pH of the last water wash was 6-7. The organic layer was washed again with brine (7.5 mL) and dried over sodium sulfate (4 g) for 20 min. The mixture was filtered into a 250 mL flask and concentrated to dryness.
The solid was dried at room temperature under high vacuum (10 mmHg or less) for 20 h.
Weight = 1.51 g of an off-white foam.
C.
Preparation of Intermediate, Compound F:
A 3 -neck 100 mL flask equipped with mechanical stirrer was charged with
2,2,5-trimethyl[l,3-dioxane]-5-carboxylic acid, Compound C (0.63 g, 3.6 mmol) in methylene chloride (7.5 mL). Dusopropylethylamine (0.77 g, 5.9 mmol) was added, followed by a rinse with methylene chloride (1 mL). 2,4,6-Trichlorobenzoyl chloride (0.85 g, 3.5 mmol) was added, followed by a rinse with methylene chloride (1.5 mL).
The mixture was held at room temperature for 4.5 h. The solution was cooled to -12 ±
2°C. 31 -Trimethylsilyl proline rapamycin, compound E, (1.51 g) in methylene chloride (8 mL) was dissolved and added to the 100 mL flask. Methylene chloride (2 mL) was added as a rinse. A solution of dimethylamino pyridine (DMAP) (0.77 g, 6.8 mmol) in methylene chloride (3 mL) was prepared and added to the 100 mL flask over
2.5 h maintaining the temperature -12 ± 2 °C. Methylene chloride (1 mL) was added as a rinse. The mixture was held for 16 h and was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL water and 0.2 mL ethyl acetate. The HPLC sample was prepared by withdrawing 2 drops of the upper organic layer, blowdrying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04
Wavelength : λ = 280 nm Flow rate : 1 mL / min
Time : 60 min
Retention times : Compound E ~14.0-14.5 min Compound F -33.4-33.8 min
The reaction was complete when < 0.5% of starting material was present. The reaction mixture was quenched with water (6 mL). Methylene chloride (10 mL) was added and the layers separated. The aqueous layer was extracted with methylene chloride (10 mL). The combined organic layers were washed with 0.5 N sulfuric acid (12 mL), brine (10 mL), saturated sodium bicarbonate (6 mL), and water (3 x 10 mL) in that order. The pH of the last water wash was 6-7. The clear yellow solution was concentrated to a foam. The solid was dried at room temperature under high vacuum (10 mmHg or less) for 24 h. Weight = 1.88 g of a yellow foam.
D.
Preparation of crude proline CCI-779
A 1-neck 50 mL flask equipped with mechanical stirrer was charged with Compound F in THF (18.8 mL, 10 vols) and then cooled to 0 - 5 °C (or about -2.5°C). 2 N sulfuric acid (9.4 mL, 5 vols) was added over 2.5 h. After complete addition, the mixture was warmed to 2.5 °C and then held for 45 h. The reaction was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 0.25 mL ethyl acetate. The HPLC sample was prepared by withdrawing 5 drops of the upper organic layer, blow drying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04 Wavelength : λ= 280 nm Flow rate : 1 mL / min Time : 60 min Retention times Compound F ~33.4-33.8 min Desilylated Compound F ~10.5-11.5 min (intermediate) Proline CCI-779 -5.0-5.5 min The desilylated intermediate of compound F was formed first. The reaction was complete when < 0.5% of the silylated analog remained. Ethyl acetate (27 mL) and brine (7.5 mL) was added and the layers separated. The aqueous layer was extracted with ethyl acetate (10 mL). The combined organic layers were washed with brine (10 mL), saturated sodium bicarbonate (7.5 mL), and water (3 x 7.5 mL) in that order. The pH of the last water wash was 6-7. The mixture was dried over sodium sulfate (5 g) for 30 min, filtered into a 250 L flask and concentrated to dryness. Weight = 1.58 g of a yellow foam.
E.
Chromatographic purification of crude proline CCI-779
A silica gel column (31.6 g, 60 A, 200-400 mesh) (22 cm length x 2.5 cm diameter) was prepared and conditioned with 15:85 acetone:HPLC grade hexane (1 L). The yellow crude proline CCI-779 (1.58 g) in acetone (1.58 mL) was prepared and chromatographed. The column was eluted with the remaining 15:85 acetone :hexane mixture followed by 25:75 acetone:hexane (4 L). The positive fractions were combined and concentrated to dryness. The resulting foam was dried at 35 °C, high vacuum (i.e., 10 mmHg or less) for 24 h. Weight = 1.12 g of a light yellow foam.
F.
Ether treatment of proline CCI-779
A 1 -neck 50 mL flask was charged with proline CCI-779 ( 1.12 g) and dissolved in ether (1.5 mL). The mixture was held for 2 h. The ether was stripped to give a foam. The foam was dried at 35 °C, under high vacuum (10 mmHg or less) for 12 h then at room temperature overnight (12 h). Weight = 1.09 g.
*H NMR (500 and 600 MHz, DMSO-d6) δ 5.45 (H-l), 6.12 (H-2), 6.27 (H-3), 6.41 (H-4), 6.20 (H-5), 3.66 (H-7), 1.14 and 1.86 (H-8), 4.02 (H-9), 1.19 and 1.81 (H-10), 1.52 (H-11), 2.03 (H-12), 3.23 and 3.54 (H-18), 1.76 (H-19), 2.20 and 1.89 (H-21), 4.22 (H-22), 4.87 (H-25), 2.28 and 2.70 (H-26), 3.22 (H-28), 5.11 (H-29), 4.04 (H-31), 4.17 (H-32), 2.25 (H-34), 0.985 and 1.38 (H-35), 2.22 (H-36), 1.76 (H-37), 0.961 and 1.11 (H-38), 1.31 (H-39), 0.726 and 1.90 (H- 40), 3.14 (H-41), 4.46 (H-42), 1.22 and 1.81 (H-43), 0.888 and 1.60 (H-44), 1.60 (H-45), 3.05 (H-46, OCH3), 0.697 (H-47), 6.48 (H-48), 0.821 (H-49), 1.76 (H-50), approx. 5.1- 5.3 (H-51), 3.17 (H-52, OCH3), 0.755 (H-53), 0.966 (H-54), 0.805 (H-55), 3.29 (H-56, OCH3), 3.46 (H-59), 1.01 (H-60), approx. 4.3-4.7 (0-61)
13C NMR (75 MHz, DMSO- d6) δ 139.12 (C-1), 130.53 (C-2), 132.49 (C-3), 127.08 (C-4), 127.21 (C-5), 137.12 (C-6), 81.93 (C-7), 40.40 (C-8), 65.83 (C-9), 29.45 (C-10), 25.87 (C-l l), 34.21 (C-12), 99.25 (C-13), 198.17 (C-15), 165.55 (C-16), 47.01 (C-18), 24.04 (C-19), 28.93 (C-21), 58.50 (C-22), 170.44 (C-23), 73.24 (C-25), 39.96 (C-26), 207.67 (C-27), 44.51 (C-28), 123.92 (C-29), 136.56 (C-30), 75.84 (C-31), 84.86 (C-32), 209.49 (C-33), 40.76 (C-34), 39.20 (C-35), 35.05 (C-36), 32.73 (C-37), 38.42 (C-38), 32.06 (C-39), 36.01 (C-40), 80.12 (C- 41), 75.92 (C-42), 29.25 (C-43), 30.24 (C-44), 10.27 (C-45), 55.48 (C-46, OCH3), 15.46 (C-47), 15.59 (C-49), 14.41 (C-50), 56.56 (C-52, OCH3), 12.67 (C-53), 21.50 (C-54), 14.89 (C-55), 57.27 (C-56, OCH3), 174.22 (C-57), 49.90 (C-58), 63.59 and 63.98 (C-59), 16.82 (C-60). MS [M+NH ] 1033.5, [ESI(+), M+Na+] 1038.7.
Example 3 - Synthesis of CCI-779:

A. Synthesis of CCI-779 via intermediate A Method 1 : A mixture of rapamycin (6 g), vinyl ester I (2 g), lipase PS-C "Amano" II (6 g) in anhydrous TBME (36 mL) was heated at 45 °C under Ar2 for 2 days. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, the oily residue was added to heptane while stirring. The batch was then cooled to -15 °C for 2 h, collect the solid on the Buchner funnel and washed with cold heptane, A was obtained as off-white solid, crude yield : 98%.MS (El): 1070 Above crude A (6g), dissolved in n-PrOH (24 mL) cooled to 0 °C with an ice-water bath, to this solution was added aqueous H2S04 (12 mL, 1.2N). The mixture was stirred for 24 h at 0°C and was then added to cold phosphate buffer (300 ml, pH=7.8), collect the solid on a Buchner funnel and washed with DI water and dry under vacuum, silica gel column purification eluting with hexane-acetone furnished CCI-779 as a white solid (5.2 g, 90%). MS (El): 1030 Method 2: A mixture of rapamycin (30.0 g, 32.8 mmol), vinyl ester I (10.0 g, 50 mmol), lipase PS-C "Amano" II (30 g) and molecular sieves (5 A) (10.0 g) in anhydrous TBME (150 mL) was heated at 42-43 °C under Ar2 for 48 hours. THF (100 mL) was added to dissolve the precipitation and the mixture was cooled to room temperature. Enzyme was removed by filtration and washed with THF (200 mL), the filtrate was concentrated to about 60 mL and diluted with THF (320 mL). The solution was then cooled to 0-5 °C, H2S04 (180 mL, 2N) was added dropwise over lh. The mixture was stirred for 48 h at 0-5 °C or until the disappearance of A as monitored by TLC. The mixture was diluted with brine (300 mL) and extracted with EtOAc (three times). The combined organic layer was washed with H20, 5% NaHC03, then brine and dried
(MgS04). Evaporation of solvent gave a light yellowish semi solid which was purified by flash chromatography (hexane/acetone, 2:1) to give CCI-779 as a white solid (30.77 g, 91% for two steps). B. Synthesis of CCI-779 via intermediate B: A mixture of rapamycin (3 g), vinyl ester II (1.2 g), lipase PS-C "Amano" II (5 g) in anhydrous TBME (45 mL) was heated at 45 °C under Ar2 for 60 h. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, MeOH (20 mL) was added to the residue and concentrated to dryness. Silica gel column purification of crude eluting with hexane-acetone furnished CCI-779 as a white solid (2.3 g), and recovered rapamycin (0.81 g). The yield is 93% based on the recovered rapamycin.
proline analog of CCI-779 (proline-rapamycin42-ester with 2,2-bis(hydroxymethyl)propionic acid or proline-CCI-779) and methods of synthesizing same. Proline-CCI-779 is an active drug substance useful in oncology and other associated indications (immunosuppression, anti-inflammatory, anti-proliferation and anti-tumor). In one aspect, the synthesis of proline-CCI-779 is accomplished through bis- silylation of proline rapamycin, mono-de-protecting 31 ,42-bis-trimethylsilyl proline rapamycin, and acylating the mono-silyl proline rapamycin followed by hydrolysis. In another aspect, the invention provides a two-step enzymatic process involving a regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to give CCI-779.
Example 4 - Synthesis of Proline-CCI-779 The enzymatic procedure of the invention can also be applied to the synthesis of proline CCI-779 from proline-rapamycin under essentially the same conditions as described in Example 2, procedure A for the synthesis of CCI-779 from rapamycin.

proline-rapamycin proline-CCI-779
......................
more info added for readers
synthesis of CCI-779 or Proline CCI-779 (Temsirolimus) which is useful as an antineoplastic agent having the structure

It is stated to be effective in multiple applications, including inhibition of tumor growth, the treatment for multiple sclerosis and rheumatoid arthritis.
2. The Prior Arts

U.S. Pat. No. 7,202,256 disclosed methods for the synthesis of CCI-779 (Temsirolimus), providing two-step enzymatic process involving regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to obtain the CCI-779 (as shown in scheme 1). A number of drawbacks of the synthesis route depicted in scheme 1 are high-priced PdCl2 and poisonous trimethylboroxine.
U.S. Pat. No. 7,202,256 disclosed methods for the synthesis of CCI-779 (Temsirolimus), providing two-step enzymatic process involving regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to obtain the CCI-779 (as shown in scheme 1). A number of drawbacks of the synthesis route depicted in scheme 1 are high-priced PdCl2 and poisonous trimethylboroxine.


A selective synthesis of 42-monoacylated product was previously conducted by reacting rapamycin 31,42-bis-silyl ether, and then the 42-sily ether protection group is selectively removed to provide rapamycin-OH-31-sily ether (U.S. Pat. No. 5,563,145). In addition, a regioselective process for the preparation of CCI-779 is also described in U.S. Pat. No. 6,277,983 (Scheme2). First, rapamycin (compound 4b) is treated with excess chlorotrimethylsilane to form rapamycin31,42-bis-trimethylsilyl ether (compound 5), and then 42-trimethylsilyl ether protection group is selectively removed in mild acid to provide rapamycin 42-OH-31-trimethylsilyl ether (compound 6). This free 42-OH was then acylated with 2,4,6-trichlorobenzyl mixed anhydride of 2,2,5-trimethyl[1,3-dioxane]-5-carboxylic acid (compound 7) at −15° C. for 16 h to give rapamycin 31-trimethylsilyl ether 42-ester (compound 8). Following treatment with mild acid for a certain period, CCI-779 can be isolated. 2,4,6-trichlorobenzyl chloride is irritant, moisture sensitive and costly.

Further, as below-depicted in Scheme 3, U.S. Pat. No. 7,153,957 disclose another method for the CCI-779. It can be prepared by the acylation of 31-silyl ether of rapamycin with the anhydride derived from the 2-phenylboronate acid to give rapamycin 31-silyl ether, 42-boronate. Thereafter, it is hydrolyzed under mild acid condition to form rapamycin 42-ester boronate. After being treated with a suitable diol, CCI-779 was obtained (Scheme 3). Mixed anhydride is not satisfactory for commercial scale synthesis because it can be kept stable only for 48 hr at −5˜0° C., not durable for longer time.
synthesis ofTemsirolimus in a more economic way.

..............
PAPERS
CCI-779
Drugs Fut 2002, 27(1): 7
Drugs Fut 2002, 27(1): 7
Organic Letters, 2005 , vol. 7, 18 pg. 3945 - 3948 seenmr
PATENTS
| United States | 5362718 | APPROVED 1994-04-18 | EXPIRY 2014-04-18 |
| Canada | 2429020 | 2009-05-26 | 2021-11-13 |
| Canada | 2187024 | 2004-08-10 | 2015-04-14 |
| US8198280 | 6-13-2012 | N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF |
| US2011281891 | 11-18-2011 | N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF |
| US7998964 | 8-17-2011 | N-Hydroxyamide Derivatives and Use Thereof |
| US7973039 | 7-6-2011 | Sulfonyl Amino Cyclic Derivatives and Use Thereof |
| US7838522 | 11-24-2010 | Benzothiazole Formulations and Use Thereof |
| US2010292231 | 11-19-2010 | Indazole Compounds for Treating Inflammatory Disorders, Demyelinating Disorders and Cancers |
| US2010249415 | 9-31-2010 | Process for preparation of temsirolimus |
| US2010098691 | 4-23-2010 | COMBINATION OF BENZIMIDAZOLE ANTI-CANCER AGENT AND A SECOND ANTI-CANCER AGENT |
| US7605257 | 10-21-2009 | Processes for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants |
| US2009149511 | 6-12-2009 | Administration of an Inhibitor of HDAC and an mTOR Inhibitor |
| US2007128731 | 6-8-2007 | Methods for preparing crystalline rapamycin and for measuring crystallinity of rapamycin compounds using differential scanning calorimetry |
| US7202256 | 4-11-2007 | Proline CCI-779, production of and uses therefor, and two-step enzymatic synthesis of proline CCI-779 and CCI-779 |
| US2007004767 | 1-5-2007 | Methods for treating neurofibromatosis 1 |
| US7074804 | 7-12-2006 | CCI-779 Isomer C |
| US5362718 | 18 Apr 1994 | 8 Nov 1994 | American Home Products Corporation | Rapamycin hydroxyesters |
| US6197967 | 13 Dec 1999 | 6 Mar 2001 | Clariant Gmbh | Process for the preparation of paraoxadiazolyphenylboronic acids |
| US6277983 | 27 Sep 2000 | 21 Aug 2001 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| WO1995028406A1 | 14 Apr 1995 | 26 Oct 1995 | American Home Prod | Rapamycin hydroxyesters, process for their preparation and pharmaceutical compositions containing them |
| US7553843 | 6 Dec 2006 | 30 Jun 2009 | Wyeth | Process for the preparation of purified crystalline CCI-779 |
| US7605258 | 16 Oct 2007 | 20 Oct 2009 | Wyeth | Processes for the synthesis of individual isomers of mono-peg CCI-779 |
| US7622578 | 6 Dec 2006 | 24 Nov 2009 | Wyeth | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US7625726 | 29 Sep 2008 | 1 Dec 2009 | Wyeth | Process for preparing rapamycin 42-esters and FK-506 32-esters with dicarboxylic acid, precursors for rapamycin conjugates and antibodies |
| US7875612 | 24 Apr 2002 | 25 Jan 2011 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US7910594 | 13 May 2003 | 22 Mar 2011 | Endocyte, Inc. | Vitamin-mitomycin conjugates |
| US8026276 | 25 Jul 2003 | 27 Sep 2011 | Wyeth Llc | Parenteral CCI-779 formulations containing cosolvents, an antioxidant, and a surfactant |
| US8044200 | 14 Mar 2006 | 25 Oct 2011 | Endocyte, Inc. | Synthesis and purification of pteroic acid and conjugates thereof |
| US8105568 | 10 Jul 2009 | 31 Jan 2012 | Endocyte, Inc. | Vitamin receptor binding drug delivery conjugates |
| US8288557 | 22 Jul 2005 | 16 Oct 2012 | Endocyte, Inc. | Bivalent linkers and conjugates thereof |
| US8299116 | 10 Aug 2011 | 30 Oct 2012 | Wyeth Llc | CCI-779 concentrate formulations |
| US8455539 | 15 Oct 2012 | 4 Jun 2013 | Wyeth Llc | CCI-779 concentrate formulations |
| US8465724 | 18 Aug 2006 | 18 Jun 2013 | Endocyte, Inc. | Multi-drug ligand conjugates |
| US8470822 | 7 May 2010 | 25 Jun 2013 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US8524893 | 28 Jan 2011 | 3 Sep 2013 | Fresenius Kabi Oncology Limited | Process for the preparation of temsirolimus and its intermediates |
| WO2011092564A2 | 20 Jan 2011 | 4 Aug 2011 | Fresenius Kabi Oncology Ltd | Process for the preparation of temsirolimus and its intermediates |