Showing posts with label US FDA. Show all posts
Showing posts with label US FDA. Show all posts
Saturday, 1 March 2014
Tuesday, 14 January 2014
Idelalisib ....US FDA Accepts NDA for Gilead’s Idelalisib for the Treatment of Refractory Indolent Non-Hodgkin’s Lymphoma

An antineoplastic agent and p110delta inhibitor
Icos (Originator)
- CAL-101
- GS-1101
- Idelalisib
- UNII-YG57I8T5M0
M.Wt: 415.43
Formula: C22H18FN7O
Formula: C22H18FN7O
CAS No.: 870281-82-6
CAL-101 Solubility: DMSO ≥80mg/mL Water <1.2mg/mL Ethanol ≥33mg/mL
CAL-101 Solubility: DMSO ≥80mg/mL Water <1.2mg/mL Ethanol ≥33mg/mL
5-Fluoro-3-phenyl-2-[(1S)-1-(7H-purin-6-ylamino)propyl]-4(3H)-quinazolinone
Idelalisib (codenamed GS-1101 or CAL-101) is a drug under investigation for the treatment of chronic lymphocytic leukaemia. It is in Phase III clinical trials testing drug combinations with rituximab and/or bendamustine as of 2013. The substance acts as aphosphoinositide 3-kinase inhibitor; more specifically, it blocks P110δ, the delta isoform of the enzyme phosphoinositide 3-kinase.[1][2]
GDC-0032 is a potent, next-generation beta isoform-sparing PI3K inhibitor targeting PI3Kα/δ/γ with IC 50 of 0.29 nM/0.12 nM/0.97nM,> 10 fold over Selective PI3K [beta].
GS-1101 is a novel, orally available small molecule inhibitor of phosphatidylinositol 3-kinase delta (PI3Kdelta) develop by Gilead and is waiting for registration in U.S. for the treatment of patients with indolent non-Hodgkin's lymphoma that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy and for the treatment of chronic lymphocytic leukemia. The compound is also in phase III clinical evaluation for the treatment of elderly patients with previously untreated small lymphocytic lymphoma (SLL) and acute myeloid leukemia. Clinical trials had been under way for the treatment of inflammation and allergic rhinitis; however, no recent development has been reported. Preclinical studies have shown that GS-1101 has desirable pharmaceutical properties. The compound was originally developed by Calistoga Pharmaceuticals, acquired by Gilead on April 1, 2011.
clinical trials, click link
FOSTER CITY, Calif.--(BUSINESS WIRE)--Jan. 13, 2014-- Gilead Sciences, Inc. (Nasdaq: GILD) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the company’s New Drug Application (NDA) for idelalisib, a targeted, oral inhibitor of PI3K delta, for the treatment of refractory indolent non-Hodgkin’s lymphoma (iNHL). FDA has granted a standard review for the iNHL NDA and has set a target review date under the Prescription Drug User Fee Act (PDUFA) of September 11, 2014.
The NDA for iNHL, submitted on September 11, 2013, was supported by a single arm Phase 2 study (Study 101-09) evaluating idelalisib in patients with iNHL that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy. Following Gilead’s NDA submission for iNHL, FDA granted idelalisib a Breakthrough Therapy designation for relapsed chronic lymphocytic leukemia (CLL). The FDA grants Breakthrough Therapy designation to drug candidates that may offer major advances in treatment over existing options. Gilead submitted an NDA for idelalisib for the treatment of CLL on December 6, 2013.
About Idelalisib
Idelalisib is an investigational, highly selective oral inhibitor of phosphoinositide 3-kinase (PI3K) delta. PI3K delta signaling is critical for the activation, proliferation, survival and trafficking of B lymphocytes and is hyperactive in many B-cell malignancies. Idelalisib is being developed both as a single agent and in combination with approved and investigational therapies.
Gilead’s clinical development program for idelalisib in iNHL includes Study 101-09 in highly refractory patients and two Phase 3 studies of idelalisib in previously treated patients. The development program in CLL includes three Phase 3 studies of idelalisib in previously treated patients. Combination therapy with idelalisib and GS-9973, Gilead’s novel spleen tyrosine kinase (Syk) inhibitor, also is being evaluated in a Phase 2 trial of patients with relapsed or refractory CLL, iNHL and other lymphoid malignancies.
Additional information about clinical studies of idelalisib and Gilead’s other investigational cancer agents can be found at www.clinicaltrials.gov. Idelalisib and GS-9973 are investigational products and their safety and efficacy have not been established.
About Indolent Non-Hodgkin’s Lymphoma
Indolent non-Hodgkin’s lymphoma refers to a group of largely incurable slow-growing lymphomas that run a relapsing course after therapy and can lead ultimately to life-threatening complications such as serious infections and marrow failure. Most iNHL patients are diagnosed at an advanced stage of disease, and median survival from time of initial diagnosis for patients with the most common form of iNHL, follicular lymphoma, is 8 to 10 years. The outlook for refractory iNHL patients is significantly poorer.
About Gilead Sciences
Gilead Sciences is a biopharmaceutical company that discovers, develops and commercializes innovative therapeutics in areas of unmet medical need. The company’s mission is to advance the care of patients suffering from life-threatening diseases worldwide. Headquartered in Foster City, California, Gilead has operations in North and South America, Europe and Asia Pacific.
The delta form of PI3K is expressed primarily in blood-cell lineages, including cells that cause or mediate hematologic malignancies, inflammation, autoimmune diseases and allergies. By specifically inhibiting only PI3K delta, a therapeutic effect is exerted without inhibiting PI3K signalling that is critical to the normal function of healthy cells. Extensive studies have shown that inhibition of other PI3K forms can cause significant toxicities, particularly with respect to glucose metabolism, which is essential for normal cell activity.
In 2011, orphan drug designation was assigned to GS-1101 in the U.S. for the treatment of CLL. In 2013, several orphan drug designations were assigned to the compound in the E.U. and U.S.: for the treatment of follicular lymphoma, for the treatment of mucosa-associated lymphoid tissue lymphoma (MALT), for the treatment of nodal marginal zone lymphoma, for the treatment of splenic marginal zone lymphoma, and for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma. Orphan drug designation was also assigned in the U.S. for the treatment of lymphoplasmacytic lymphoma with or without Walenstom's macroglobulinemia and, in the E.U., for the treatment of Waldenstrom's macroglobulinemia (lymphoplasmacytic lymphoma).
Later in 2013, some of these orphan drug designations were withdrawn in the E.U.; for the treatment of chronic lymphocytic leukemia / small lymphocytic lymphoma, for the treatment of extranodal marginal-zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma), for the treatment of of nodal marginal-zone lymphoma and for the treatment of splenic marginal-zone lymphoma. In 2013, the FDA granted a breakthrough therapy designation for the treatment of chronic lymphocytic leukemia.
- H. Spreitzer (13 May 2013). "Neue Wirkstoffe – Ibrutinib und Idelalisib". Österreichische Apothekerzeitung (in German) (10/2013): 34.
- Wu, M.; Akinleye, A.; Zhu, X. (2013). "Novel agents for chronic lymphocytic leukemia".Journal of Hematology & Oncology 6: 36. doi:10.1186/1756-8722-6-36.PMC 3659027. PMID 23680477.
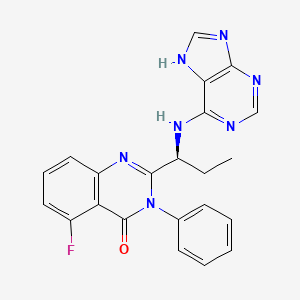 idelalisib
idelalisibCAL-101 is an Oral Delta Isoform-Selective PI3 Kinase Inhibitor.
CAL-101 (GS 1101) is a potent PI3K p110δ inhibitor with an IC50 of 65 nM. PI3K-delta inhibitor CAL-101 inhibits the production of the second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3), preventing the activation of the PI3K signaling pathway and thus inhibiting tumor cell proliferation, motility, and survival. Unlike other isoforms of PI3K, PI3K-delta is expressed primarily in hematopoietic lineages. The targeted inhibition of PI3K-delta is designed to preserve PI3K signaling in normal, non-neoplastic cells. [3][4]
Reference:
[3] Blood 2011, 117, 591-594.
[4] Blood, 2010, 116, 2078-2088.
5. WO 2005113556
6. WO 2005113554
7. WO 2010057048
8. WO 2011156759
9. WO 2012125510
10. WO 2013134288
11. US 2013274198
12. J Med Chem. 2013 Mar 14;56(5):1922-39. doi: 10.1021/jm301522m
| US8207153 | 6-27-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2012015964 | 1-20-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2011306622 | 12-16-2011 | METHODS OF TREATING HEMATOLOGICAL DISORDERS WITH QUINAZOLINONE COMPOUNDS IN SELECTED SUBJECTS |
| US7932260 | 4-27-2011 | Quinazolinones as Inhibitors of Human Phosphatidylinositol 3-Kinase Delta |
| US2011044942 | 2-25-2011 | METHODS OF TREATMENT FOR SOLID TUMORS |
| US2010256167 | 10-8-2010 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2010202963 | 8-13-2010 | THERAPIES FOR HEMATOLOGIC MALIGNANCIES |
| WO2005113556A1 * | 12 May 2005 | 1 Dec 2005 | Icos Corp | Quinazolinones as inhibitors of human phosphatidylinositol 3-kinase delta |
| WO2005117889A1 * | 12 Nov 2004 | 15 Dec 2005 | Didier Bouscary | Methods for treating and/or preventing aberrant proliferation of hematopoietic |
| WO2005120511A1 * | 4 Jun 2005 | 22 Dec 2005 | Joel S Hayflick | Methods for treating mast cell disorders |
| WO2006089106A2 * | 16 Feb 2006 | 24 Aug 2006 | Icos Corp | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| US20060106038 * | 25 May 2005 | 18 May 2006 | Icos Corporation | Methods for treating and/or preventing aberrant proliferation of hematopoietic cells |
............................
synthesis
The synthesis of a compound in accordance with formula I is first exemplified using steps A-E below, which provide a synthetic procedure for compound 107, the structure of which is shown below.

(107) is idelalisib
...................
Synthesis of 2-fluoro-6-nitro-N-phenyl-benzamide (108)
Step A: A solution of 2-fluoro-6- nitrobenzoic acid (100 g, 0.54 mol) and dimethylformamide (5 mL) in dichloromethane (600 mL) was treated dropwise with oxalyl chloride (2 M in dichloromethane, 410 mL, 0.8 mol, 1.5 eq) over 30 min. After stirring 2 h at room temperature, the reaction was concentrated to an orange syrup with some solids present. The syrup was dissolved in dry dioxane (80 mL) and slowly added to a suspension of aniline (49 mL, 0.54 mol, 1 eq) and sodium bicarbonate (90 g, 1.08 mol, 2 eq) in a mixture of dioxane (250 mL) and water (250 mL) at 6 0C. The temperature reached 27°C at the end of the addition. After 30 min, the reaction mixture was treated with water (1.2 L). The precipitate was collected by vacuum filtration, washed with water (300 mL) , air dried in the funnel, and dried in vacuo at 50°C for 24 h to afford an off-white solid product (139 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 10.82 (s, IH), 8.12 (d, J = 7.7 Hz, IH), 7.91-7.77 (m, 2H), 7.64 (d, J = 7.7 Hz, 2H), 7.38 (t, J = 7.9 Hz, 2H), 7.15 > (t, J = 7.4 Hz, IH), ESI-MS m/z 261 (MH+). The reaction described above and compound 108 are shown below.

.............................
Synthesis of(S) - [1- (2-fluoro-6-nitro-benzoyl) -phenyl-aminocarbonyl] - propyl-carbamic acid tert-butyl ester (109)
Step B: A suspension of compound 108 (0.5 mol) and dimethylformamide (5 mL) in thionyl chloride (256 mL, 2.5 mol, 5 eq) was stirred at 85°C for 5 hours. The reaction mixture was concentrated in vacuo to a brown syrup. The syrup was dissolved in dichloromethane (200 mL) and was slowly added to a solution of N-BOC-L-2-aminobutyric acid (112 g, 0.55 mol, 1.1 eq) and triethylamine (77 mL, 0.55 mol, 1.1 eq) in dichloromethane (600 mL) at 10 0C. After stirring at room temperature for 3 h, salts were removed by filtration, and the solution was washed with 100 mL of water, saturated sodium bicarbonate, water, 5% citric acid, and saturated sodium chloride. The organic phase was dried with magnesium sulfate and concentrated to a red syrup. The syrup was dissolved in dichloromethane (450 mL) and purified by flash chromatography on a silica gel plug (15 x 22 cm, 4 L dry silica) eluted with hexanes/ethyl acetate (10%, 8 L; 15%, 8 L; 20%, 8 L; 25%, 4 L) to yield the compound 109 as an off-white solid (147 g, 66%). 1H NMR (300 MHz, DMSO-d6) δ 8.13 (d, J = 8.0 Hz, IH), 7.84 (t, J = 8.6 Hz, IH), 7.78- 7.67 (m, IH), 7.65-7.49 (m, 3H), 7.40-7.28 ( m, 2H), 7.19 (d, J = 7.5 Hz, IH), 4.05 (broad s, IH), 1.75- 1.30 (m, 2H), 1.34 (s, 9H), 0.93 (broad s, 3H). ESI- MS m/z 446.3 (MH+) . The reaction described above and compound 109 are shown below.

.........................
Synthesis of(S) - [1- (5-fluoro-4-oxo-3-phenyl-3 , 4-dihydro-quinazolin-2- yl) -propyl] -carbamic acid tert-butyl ester (110)
Step C: A solution of compound 109 (125 mmol, 1 eq) in acetic acid (500 mL) was treated with zinc dust (48.4 g, 740 mmol, 6 eq) added in 3 portions, and the reaction mixture was allowed to cool to below 35°C between additions. After stirring for 2 h at ambient temperature, solids were filtered off by vacuum filtration and washed with acetic acid (50 mL) . The filtrate was concentrated in vacuo, dissolved in EtOAc (400 mL) , washed with water (300 mL) , and the water layer was extracted with EtOAc (300 mL) . The combined organic layers were washed with water (200 mL) , sat'd sodium bicarbonate (2 x 200 mL) , sat'd NaCl (100 mL) , dried with MgSO4, and concentrated to a syrup. The syrup was dissolved in toluene (200 mL) and purified by flash chromatography on a silica gel plug (13 x 15 cm, 2 L dry silica) eluted with hexanes/ethyl acetate (10%, 4 L; 15%, 4 L; 17.5%, 8 L; 25%, 4 L) to yield compound 110 as an off-white foamy solid (33.6 g, 69%). 1H NMR (300 MHz, DMSO-d6) δ 7.83 (td, J = 8.2, 5.7 Hz, IH), 7.64-7.48 (m, 5H), 7.39 (broad d, J = 7.6 Hz, IH), 7.30 (dd, J = 8.3 Hz, IH), 7.23 (d, J = 7.6 Hz, IH), 4.02-3.90 (m, IH), 1.76-1.66 (m, IH), 1.62-1.46 (m, IH), 1.33 (s, 9H), 0.63 (t, J= 7.3 Hz, 3H). ESI-MS m/z 398.3 (MH+). The reaction described above and compound 110 are shown below.

..............
Syn of (S) -2- (1-amino-propyl) -5-fluoro-3-phenyl-3H-quinazolin-4- one (111)
Step D: A solution of compound 110 (85 mmol) in dichloromethane (60 mL) was treated with trifluoroacetic acid (60 mL) . The reaction mixture was stirred for 1 h, concentrated in vacuo, and partitioned between dichloromethane (150 mL) and 10% K2CO3 (sufficient amount to keep the pH greated than 10) . The aqueous layer was extracted with additional dichloromethane (100 raL) , and the combined organic layers were washed with water (50 mli) and brine (50 mL) . After drying with Mg SO4, the solution was concentrated to provide compound 111 as an off-white solid (22 g, 88%) . 1H NMR (300 MHz,
CDCl3) δ 7.73-7.65 (m, IH), 7.62-7.49 (m, 4H), 7.32- 7.22 (m, 2H), 7.13-7.06 (m, IH), 3.42 (dd, J= 7.5, 5.2 Hz, IH), 1.87-1.70 (m, IH), 1.58-1.43 (m, IH), 0.80 (t, J = 7.4 Hz, 3H) . ESI-MS m/z 298.2 (MH+) . The reaction described above and compound 111 are shown below.

..................
syn of (S) -5-fluoro-3-phenyl-2- [1- (9H-purin-6-ylamino) -propyl] - 3H-quinazolin-4-one (107)
Step E: A suspension of compound 111(65.6 mmol, 1 eq) , 6-bromopurine (14.6 g, 73.4 mmol, 1.1 eq) , and DIEA (24.3 mL, 140 mmol, 2 eq) in tert- butanol (40 mL) was stirred for 24 h at 800C. The reaction mixture was concentrated in vacuo and treated with water to yield a solid crude product that was collected by vacuum filtration, washed with water, and air dried. Half of the obtained solid crude product was dissolved in MeOH (600 mL) , concentrated onto silica gel (300 mL dry) , and purified by flash chromatography (7.5 x 36 cm, eluted with 10 L of 4% MeOH/CH2Cl2) to yield a solid product. The solid product was then dissolved in EtOH (250 mL) and concentrated in vacuo to compound 107 idelalisib as a light yellow solid (7.2 g, 50%).
1H NMR (300 MHz, 80 0C, DMSO-d5) δ 12.66 (broad s, IH), 8.11 (s, IH), 8.02 (broad s, IH), 7.81-7.73 (m, IH),7.60-7.42 (m, 6H), 7.25-7.15 (m, 2H), 4.97 (broad s, IH), 2.02-1.73 (m, 2H), 0.79 (t, J= 7.3 Hz, 3H).
ESI-MS m/z 416.2 (MH+).
C, H, N elemental analysis (C22Hi8N7OF-EtOH- 0.4 H2O).
Chiral purity 99.8:0.2 (S:R) using chiral HPLC (4.6 x 250 mm Chiralpak ODH column, 20 °C, 85:15 hexanes : EtOH, 1 rnL/min, sample loaded at a concentration of 1 mg/mL in EtOH) . The reaction described above and compound 107 idelalisib are shown below.

| WO2001030768A1 * | 26 Oct 2000 | 3 May 2001 | Gustave Bergnes | Methods and compositions utilizing quinazolinones |
| WO2001081346A2 * | 24 Apr 2001 | 1 Nov 2001 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2003035075A1 * | 27 Aug 2002 | 1 May 2003 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2005016348A1 * | 13 Aug 2004 | 24 Feb 2005 | Jason Douangpanya | Method of inhibiting immune responses stimulated by an endogenous factor |
| WO2005016349A1 * | 13 Aug 2004 | 24 Feb 2005 | Thomas G Diacovo | Methods of inhibiting leukocyte accumulation |
| WO2005067901A2 * | 7 Jan 2005 | 28 Jul 2005 | Carrie A Northcott | Methods for treating and preventing hypertension and hypertension-related disorders |
Subscribe to:
Posts (Atom)