
DEGARELIX
214766-78-6 (free base)
214766-78-6 (free base)
Degarelix Acetate , 934246-14-7 cas
NICE backs Ferring's Firmagon for prostate cancer subgroup
Published on Friday, December 27, 2013
The U.K.'s NICE issued draft guidance recommending Firmagon degarelix from Ferring Pharmaceuticals A/S (Saint-Prex, Switzerland) to treat advanced hormone-dependent prostate cancer only in a subgroup of patients with spinal metastases who are at risk of impending spinal cord compression. The gonadotropin-releasing hormone (GnRH) antagonist is approved in the EU to treat advanced hormone-dependent prostate cancer. NICE said the incremental cost-effectiveness ratios (ICERs) per quality-adjusted life year (QALY) for Firmagon compared to luteinizing hormone-releasing hormone (LHRH) agonists -- which are used as first-line treatment for hormone-dependent prostate cancer -- were L26,200-L103,200 ($42,876-$168,887), all outside the range normally considered cost-effective. However, the committee said Firmagon provided a benefit and was cost-effective for the subgroup of patients with spinal metastasis, for which there are no available treatments

FIRMAGON is a sterile lyophilized powder for injection containing degarelix (as the acetate) and mannitol. Degarelix is a synthetic linear decapeptide amide containing seven unnatural amino acids, five of which are D-amino acids. The acetate salt of degarelix is a white to off-white amorphous powder of low density as obtained after lyophilization.
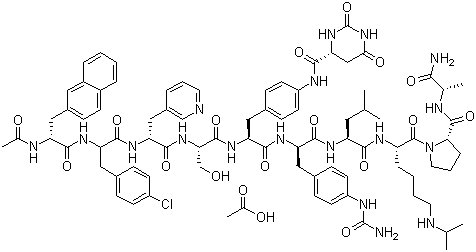
The chemical name of degarelix is D-Alaninamide, N-acetyl-3-(2-naphthalenyl)-D-alanyl-4-chloro-Dphenylalanyl-3-(3-pyridinyl)-D-alanyl-L-seryl-4-[[[(4S)-hexahydro-2,6-dioxo-4pyrimidinyl]carbonyl]amino]-L phenylalanyl-4-[(aminocarbonyl)amino]-D-phenylalanyl-L leucyl-N6–(1-methylethyl)-L-lysyl-L-prolyl. It has an empirical formula of C82H103N18O16Cl and a molecular weight of 1632.3 Da.
Degarelix has the following structural formula:
 |
FIRMAGON delivers degarelix acetate, equivalent to 120 mg of degarelix for the starting dose, and 80 mg of degarelix for the maintenance dose. The 80 mg vial contains 200 mg mannitol and the 120 mg vial contains 150 mg mannitol.
........................................
Degarelix (INN) or degarelix acetate (USAN) (tradename Firmagon) is a hormonal therapy used in the treatment of prostate cancer. During development it was known as FE200486.
Testosterone is a male hormone that promotes growth of many prostate tumours and therefore reducing circulating testosterone to very low (castration) levels is often the treatment goal in the management of men with advanced prostate cancer. Degarelix has an immediate onset of action, binding to gonadotropin-releasing hormone (GnRH) receptors in the pituitary gland and blocking their interaction with GnRH. This induces a fast and profound reduction in luteinising hormone (LH), follicle-stimulating hormone (FSH) and in turn, testosterone suppression.[1]

On 24 December 2008, the Food and Drug Administration (FDA) approved degarelix for the treatment of patients with advanced prostate cancer in the USA.[2] It was subsequently approved by the European Commission at the recommendation of the European Medicines Agency (EMEA) on February 17, 2009 for use in adult male patients with advanced, hormone-dependent prostate cancer. Ferring Pharmaceuticals markets the drug under the name Firmagon.
GnRH antagonists (receptor blockers) such as degarelix are a new type of hormonal therapy for prostate cancer. These agents are synthetic peptidederivatives of the natural GnRH decapeptide – a hormone that is made by neurons in the hypothalamus. GnRH antagonists compete with natural GnRH for binding to GnRH receptors in the pituitary gland. This reversible binding blocks the release of LH and FSH from the pituitary. The reduction in LH subsequently leads to a rapid and sustained suppression of testosterone release from the testes and subsequently reduces the size and growth of the prostate cancer. This in turn results in a reduction in prostate-specific antigen (PSA) levels in the patient's blood. Measuring PSA levels is a way to monitor how patients with prostate cancer are responding to treatment.
Unlike the GnRH agonists, which cause an initial stimulation of the hypothalamic-pituitary-gonadal axis (HPGA), leading to a surge in testosterone levels, and under certain circumstances, a flare-up of the tumour, GnRH antagonists do not cause a surge in testosterone or clinical flare.[3] Clinical flare is a phenomenon that occurs in patients with advanced disease, which can precipitate a range of clinical symptoms such as bone pain, urethralobstruction, and spinal cord compression. Drug agencies have issued boxed warnings regarding this phenomenon in the prescribing information for GnRH agonists. As testosterone surge does not occur with GnRH antagonists, there is no need for patients to receive an antiandrogen as flare protection during prostate cancer treatment. GnRH agonists also induce an increase in testosterone levels after each reinjection of the drug – a phenomenon that does not occur with GnRH antagonists such as degarelix.
GnRH antagonists have an immediate onset of action leading to a fast and profound suppression of testosterone and are therefore especially valuable in the treatment of patients with prostate cancer where fast control of disease is needed.
Clinical effectiveness
A Phase III, randomised, 12 month clinical trial (CS21) in prostate cancer[4] compared androgen deprivation with one of two doses of degarelix or the GnRH agonist, leuprolide. Both degarelix doses were at least as effective as leuprolide at suppressing testosterone to castration levels (≤0.5 ng/mL) from Day 28 to study end (Day 364). Testosterone levels were suppressed significantly faster with degarelix than with leuprolide, with degarelix uniformly achieving castration levels by Day 3 of treatment which was not seen in the leuprolide group. There were no testosterone surges with degarelix compared with surges in 81% of those who received leuprolide. Degarelix resulted in a faster reduction in PSA levels compared with leuprolide indicating faster control of the prostate cancer. Recent results also suggest that degarelix therapy may result in longer control of prostate cancer compared with leuprolide.[5]
- ^ Princivalle M, Broqua P, White R, et al (March 2007). Rapid suppression of plasma testosterone levels and tumor growth in the dunning rat model treated with degarelix, a new gonadotropin-releasing hormone antagonist. J. Pharmacol. Exp. Ther. 320: 1113-8.
- ^ PR Newswire. FDA approves Ferring Pharmaceuticals' Degarelix (generic name) for the treatment of advanced prostate cancer. PR Newswire, Europe Ltd 2008 [cited 2009 Mar 2]; Available from here
- ^ Van Poppel H, Nilsson S (June 2008). Testosterone surge: rationale for gonadotropin-releasing hormone blockers? Urology 71: 1001-6.
- ^ a b c Klotz L, Boccon-Gibod L, Shore ND, et al (December 2008). The efficacy and safety of degarelix: a 12-month, comparative, randomized, open-label, parallel-group phase III study in patients with prostate cancer. BJU Int. 102: 1531-8.
- ^ Schröder FH, Boccon-Gibod L, Tombal B, et al (March 2009) Degarelix versus leuprolide in patients with prostate cancer: effect in metastatic patients as assessed by serum alkaline phosphatase.European Association of Urology (EAU) Annual congress 17–21 March 2009, Stockholm, Sweden. Abstract 40.
- ^ Gittelman M, Pommerville PJ, Persson BE, et al (November 2008). A 1-year, open label, randomized phase II dose finding study of degarelix for the treatment of prostate cancer in North America. J. Urol. 180: 1986-92.
- ^ Van Poppel H, Tombal B, de la Rosette JJ, et al (October 2008). Degarelix: a novel gonadotropin-releasing hormone (GnRH) receptor blocker--results from a 1-yr, multicentre, randomised, phase 2 dosage-finding study in the treatment of prostate cancer. Eur. Urol. 54: 805-13.
- ^ a b Radu, A.; Pichon, C.; Camparo, P.; Antoine, M.; Allory, Y.; Couvelard, A.; Fromont, G. L.; Hai, M. T. V.; Ghinea, N. (2010). "Expression of Follicle-Stimulating Hormone Receptor in Tumor Blood Vessels". New England Journal of Medicine 363 (17): 1621–1630. doi:10.1056/NEJMoa1001283. PMID 20961245.

N-Boc-D-alanine (I) was coupled to the resin using diisopropyl carbodiimide and 1-hydroxybenzotriazole to afford resin (II). Subsequent cleavage of the Boc protecting group by means of trifluoroacetic acid provided the D-alanine-bound resin ( III) Sequential coupling and deprotection cycles were carried out with the following protected amino acids:. N-Boc-L-proline (IV), N-alpha-Boc-N6-isopropyl-N6-carbobenzoxy-L-lysine (VI) and N-Boc-L-leucine (VIII) to afford the respective peptide resins (V), (VII) and (IX). N-alpha-Boc-D-4-(Fmoc-amino) phenylalanine (X) was coupled to (IX), yielding resin (XI). Cleavage of the side-chain Fmoc protecting group with piperidine in DMF gave the aniline derivative (XII). After conversion to the corresponding urea by treatment with tert-butyl isocyanate, the Boc group was cleaved with trifluoroacetic acid to produce resin (XIII).
J Med Chem 2001, 44, 3,. 453
WO 9846634.
Drugs Fut 2006, 31(9): 755
The synthesis of Degarelix has been described by solid phase synthesis using Boc chemistry (2001 :880 SYNTHLINE):
N-Boc-D-alanine (I) was coupled to the MBHA resin using diisopropyl carbodiimide and 1-hydroxybenzotriazole to afford resin (II). Subsequent cleavage of the Boc protecting group by means of trifluoroacetic acid (TFA) provided the D-alanine-bound resin (III). Sequential coupling and deprotection cycles were carried out with the following protected amino acids: N-Boc-L-proline (IV), N-alpha-Boc-N6-isopropyl-N6-carbobenzoxy-L- lysine (VI) and N-Boc-L-leucine (VIII) to afford the respective peptide resins (V), (VII) and (IX). N-alpha-Boc-D-4-(Fmoc-amino)phenylalanine (X) was coupled to (IX), yielding resin (XI). Cleavage of the side-chain Fmoc protecting group with piperidine DMF gave the aniline derivative (XII). This portion of the synthesis route is shown below in Scheme 1.
Scheme 1

Sche

Scheme 3

Scheme 4

Scheme 5


US 6214798 Bl, which describes a similar synthetic approach, suggests that classical peptide solution synthesis would be preferable for producing large quantities of product. The purity of the final material obtained in US 6214798 Bl is estimated, therein, to be about 98%, yet J Med. Chem. 2005, 48, 4851-4860 describing the same preparation yields a purity as measured by capillary zone electrophoresis (CZE) of 98%, which corresponds to 96% according to HPLC analysis.
Fmoc chemistry generally employs milder reaction conditions than the Boc chemistry, and thus can sometimes be less likely to cause the formation of by-products..
In addition, Fmoc chemistry does not employ hydrofluoric acid or other very strong acid (such as TFMSA or HBr/AcOH) is needed. Therefore these processes are much safer and environmentally friendly; factors that are important when engineering such a synthesis process on the large production scale.
In stepwise SPPS at least two types of protection for reactive functionalities other than those involved in peptide bond formation are necessary: temporary protection of the alpha-amino group, removable after formation of each peptide bond; and, side chain protection removable after assembly of the complete peptide chain.
When using Fmoc chemistry, a tBu protecting group is commonly used for the protection of the hydroxyl groups on Ser, Tyr and Thr residues. The tBu group is easily removed together with other side-chain protecting groups at the end of the peptide synthesis by using TFA.
WO2010121835 (*W0'835') describes the preparation of Degarelix using an Fmoc strategy and using tBu as the protecting group for the Ser residue. WO'835 also teaches that, unusually severe cleavage conditions such as long reaction time and 100% TFA as cleavage cocktail were required to successfully complete the final deprotection.. Such severe conditions are commonly known to result in increased side reactions, increased degradation of the peptide, and accordingly production of a lower quality of the resulting product.
One alternative to avoid using these severe deprotection conditions could be the use of different acid labile protecting group for Ser, such as Trt. However, it is well laiown in the art that the Trt group is very bulky and hydrophobic, and is frequently used to increase .the "steric hindrance" effect. See, e.g. Chem. Lett. 27, 1999 and J Pept. Sci. 2010; 16: 364-374, which describes that using the Trt protecting group in an Fmoc peptide synthesis could prevent formation of the target peptide due to "steric hindrance." The Degarelix sequence is inherently very hydrophobic. Therefore coupling of a large hydrophobic residue to the growing peptide chain is expected to cause slowing of the reaction and thus increased liability for the formation of impurities associated with deletions in the sequence and racemization. Synthesis efficiency is an important consideration when determining which general coupling reaction to use in an SPPS process. This efficacy can be limited by a number of different variables. One major variable that must be considered is the steric hindrance. This steric hindrance is determined by the nature of the peptide side chains and of their protecting groups. It is an important variable because it will allow the rapid acylation of the amino function and have a limiting effect on possible side reactions. See, e.g., ChemPep®, Fmoc Solid Phase Peptide Synthesis; Coupling reaction). Moreover, for the Degarelix synthesis, Fmoc-
Ser(Trt)-OH would be added to extremely bulky 4Aph(L-Hor) [4-[[[(4S)-hexahydro-2,6- dioxo-4-pyrimidinyl]carbonyl] amino] -L-phenylalanyl] residue. Accordingly, the use of this protecting residue would likely be quite difficult due to "steric hindrance". These "steric effects" are known to be of great importance when two molecules or two groups are in a close approach resulting in van der Waals forces repulsive effect. The repulsive potential can become quite large if the nonbonded distances are sufficiently short. This van der Walls repulsion can thus serve to slow a reaction, a situation that is typically refen-ed to as "steric hindrance" (Stereochemistry of Organic Compounds, E.L. Eliel et al, John Willey & Sons, 1994, 720 - 721 )
patents
WO 2011004260
WO 2011066386
WO 2012013331
WO 2012055903
WO 2012055905
WO 2013104745
WO 2013178788
patents
WO 2011004260
WO 2011066386
WO 2012013331
WO 2012055903
WO 2012055905
WO 2013104745
WO 2013178788