
Plerixafor
cas 110078-46-1
CXCR4 chemokine antagonist
Stem cell mobilization [CXCR4 receptor antagonist]
A bicyclam derivate, highly potent & selective inhibitor of HIV-1 & HIV-2.
Bone marrow transplantation; Chronic lymphocytic leukemia; Chronic myelocytic leukemia; Myelodysplastic syndrome; Neutropenia; Sickle cell anemia
Plerixafor; Mozobil; AMD3100; 110078-46-1; Amd 3100; bicyclam JM-2987; AMD-3100; UNII-S915P5499N; JM3100
- JKL 169
- Mozobil
- Plerixafor
- SDZ SID 791
- UNII-S915P5499N
Molecular Formula: C28H54N8
Molecular Weight: 502.78196
1,4-
1,4,8,11-Tetraazacyclotetradecane, 1,1'-(1,4-phenylenebis(methylene))bis-

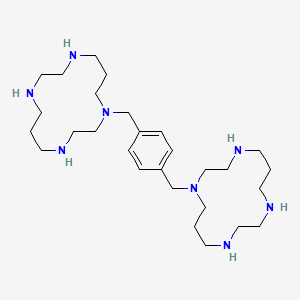
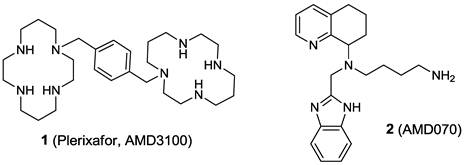
1,1′-[1,4-phenylenebis(methylene)]bis [1,4,8,11-tetraazacyclotetradecane]
1,1'- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane
Johnson Matthey (Innovator)
Plerixafor is a hematopoietic stem cell mobilizer. It is used to stimulate the release of stem cells from the bone marrow into the blood in patients with non-Hodgkin lymphoma and multiple myeloma for the purpose of stimulating the immune system. These stem cells are then collected and used in autologous stem cell transplantation to replace blood-forming cells that were destroyed by chemotherapy. Plerixafor has orphan drug status in the United States and European Union; it was approved by the U.S. Food and Drug Administration on December 15, 2008.
Mozobil (plerixafor injection) is a sterile, preservative-free, clear, colorless to pale yellow, isotonic solution for subcutaneous injection. Each mL of the sterile solution contains 20 mg of plerixafor. Each single-use vial is filled to deliver 1.2 mL of the sterile solution that contains 24 mg of plerixafor and 5.9 mg of sodium chloride in Water for Injection adjusted to a pH of 6.0 to 7.5 with hydrochloric acid and with sodium hydroxide, if required.
Plerixafor is a hematopoietic stem cell mobilizer with a chemical name l, 1'-[1,4phenylenebis (methylene)]-bis-1,4,8,11-tetraazacyclotetradecane. It has the molecular formula C28H54N8. The molecular weight of plerixafor is 502.79 g/mol. The structural formula is provided in Figure 1.
Figure 1: Structural Formula
 |
Plerixafor is a white to off-white crystalline solid. It is hygroscopic. Plerixafor has a typical melting point of 131.5 °C. The partition coefficient of plerixafor between 1octanol and pH 7 aqueous buffer is < 0.1.

Plerixafor (hydrochloride hydrate) |
(CAS 155148-31-5)
| Formal Name | 1,4- |
|---|---|
| CAS Number | 155148-31-5 |
| Molecular Formula | C28H54N8 • 8HCl • [XH2O] |
| Formula Weight | 794.5 |
The α-
Plerixafor hydrochloride (AMD-3100), a chemokine CXCR4 (SDF-1) antagonist, is launched in the U.S. for the following indications: to enhance mobilization of hematopoietic stem cells for autologous transplantation in patients with lymphoma and to enhance mobilization of hematopoietic stem cells for transplantation in patients with multiple myeloma.
In 2009, the product was approved in EU for these indications.AnorMED filed an orphan drug application for AMD-3100 with the FDA in January 2003 and received approval in July 2003 as immunostimulation for increasing the stem cells available in patients with multiple myeloma and non-Hodgkin's lymphoma. Orphan drug status was also granted by the EMEA in October 2004 as a treatment to mobilize progenitor cells prior to stem cell transplantation.
In 2011, orphan drug designation was assigned by the FDA for the treatment of AML and by the EMA for the adjunctive treatment to cytotoxic therapy in acute myeloid leukemia.
Plerixafor (rINN and USAN, trade name Mozobil) is an immunostimulant used to mobilize hematopoietic stem cells in cancer patients. The stem cells are subsequently transplanted back to the patient. The drug was developed by AnorMED which was subsequently bought by Genzyme.
History
The molecule 1,1′-[1,4-phenylenebis(methylene)]bis [1,4,8,11-tetraazacyclotetradecane], consisting of two cyclam rings linked at the amine nitrogen atoms by a 1,4-xylyl spacer, was first synthesised by Fabbrizzi et al. in 1987 to carry out basic studies on the redox chemistry of dimetallic coordination compounds.[1] Then, it was serendipitously discovered by De Clercq that such a molecule, could have a potential use in the treatment of HIV[2] because of its role in the blocking of CXCR4, a chemokine receptor which acts as a co-receptor for certain strains of HIV (along with the virus's main cellular receptor, CD4).[2]Development of this indication was terminated because of lacking oral availability and cardiac disturbances. Further studies led to the new indication for cancer patients.[3]
Indications
Peripheral blood stem cell mobilization, which is important as a source of hematopoietic stem cells for transplantation, is generally performed using granulocyte colony-stimulating factor (G-CSF), but is ineffective in around 15 to 20% of patients. Combination of G-CSF with plerixafor increases the percentage of persons that respond to the therapy and produce enough stem cells for transplantation.[4] The drug is approved for patients with lymphoma and multiple myeloma.[5]
Contraindications
Pregnancy and lactation
Studies in pregnant animals have shown teratogenic effects. Plerixafor is therefore contraindicated in pregnant women except in critical cases. Fertile women are required to use contraception. It is not known whether the drug is secreted into the breast milk. Breast feeding should be discontinued during therapy.[5]
Adverse effects
Nausea, diarrhea and local reactions were observed in over 10% of patients. Other problems with digestion and general symptoms like dizziness, headache, and muscular pain are also relatively common; they were found in more than 1% of patients. Allergies occur in less than 1% of cases. Most adverse effects in clinical trials were mild and transient.[5][6]
The European Medicines Agency has listed a number of safety concerns to be evaluated on a post-marketing basis, most notably the theoretical possibilities of spleen rupture and tumor cell mobilisation. The first concern has been raised because splenomegaly was observed in animal studies, and G-CSF can cause spleen rupture in rare cases. Mobilisation of tumor cells has occurred in patients with leukaemia treated with plerixafor.[7]
Phase III clinical development in combination with G-CSF (granulocyte colony-stimulating factor) is under way at Genzyme (which acquired the product through its acquisition of AnorMED in late 2006) in a stem cell mobilization regimen in non-Hodgkin's lymphoma (NHL). The trials are designed to evaluate the potential of plerixafor in combination with G-CSF, to rapidly increase the number of peripheral blood stem cells capable of engraftment, thereby increasing the proportion of patients reaching a peripheral blood stem cell target and, as a result, reducing the number of apheresis sessions required for patients to collect a target number of peripheral blood stem cells. A phase I safety trial had been under way for the treatment of renal cancer, however, no recent development for this indication has been reported. An IND has been filed in the U.S. seeking approval to initiate clinical evaluation of the drug candidate to help repair damaged heart tissue in patients who have suffered heart attacks. Currently, an investigator-sponsored study is ongoing to evaluate plerixafor as a single agent in allogeneic transplant. AMD-3100, in combination with mitoxantrone, etoposide and cytarabine, is also in phase I/II clinical trials at the University of Washington for the treatment of acute myeloid leukemia (AML).
The University has also been conducting early clinical trials for increasing the stem cells available for transplantation in patients with advanced hematological malignancies, however, no recent developments on this trial have been reported. Genzyme has completed a phase I/II clinical study of plerixafor hydrochloride in combination with rituximab for the treatment of chronic lymphocytic leukemia. The former AnorMED had been developing plerixafor for the treatment of rheumatoid arthritis (RA), but no clinical development has been reported as of late. AnorMED was also developing plerixafor for the treatment of HIV, but discontinued the trials in 2001 due to abnormal cardiac activity and lack of efficacy.
By blocking CXCR4, a specific cellular receptor, plerixafor triggers the rapid movement of stem cells out of the bone marrow and into circulating blood. Once in the circulating blood, the stem cells can be collected for use in stem cell transplant. In terms of use for cardiac applications, there is clinical evidence that the presence of stem cells circulating in the bloodstream or directly injected into the hearts of patients who have suffered a heart attack may result in improved cardiac function.
Chemical properties
Plerixafor is a macrocyclic compound and a bicyclam derivative.[4] It is a strong base; all eight nitrogen atoms accept protons readily. The two macrocyclic rings form chelate complexes with bivalent metal ions, especially zinc, copper and nickel, as well as cobalt and rhodium. The biologically active form of plerixafor is its zinc complex.[8]
Synthesis
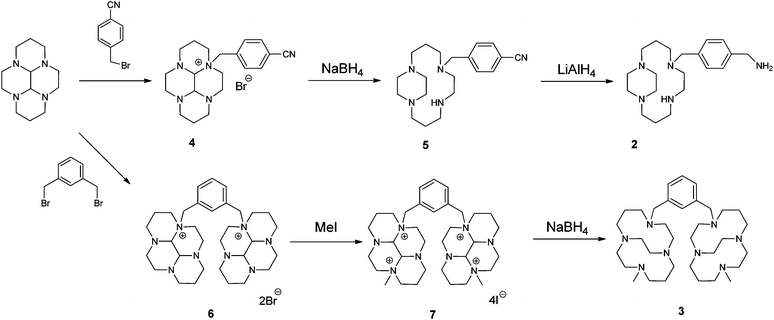
Three of the four nitrogen atoms of the macrocycle 1,4,8,11-tetraazacyclotetradecan are protected with tosyl groups. The product is treated with 1,4-dimethoxybenzene or 1,4-bis(brommethyl)benzene and potassium carbonate in acetonitrile. After cleaving of the tosyl groups with hydrobromic acid, plerixafor octahydrobromide is obtained.[9]
SEE CHINESE JOURNAL OF MEDICINAL CHEMISTRY 2010 20 (6): 511-513 ISSN: 1005-0108 CN: 21-1313/R
DOWNLOAD.........http://download.bioon.com.cn/upload/201207/24113552_9395.pdf
( 1 ) BASE FORM
0155g ( 8016% ), m p 129 ~ 131 e 。
1H-NM R
( CDC l3 ) D: 7.28( s, 4H, A r-H ), 3.55 ( br s, 4H,A r-CH2 ), 2.82 ~ 2.52( m, 32H, NCH2, NHCH2 ),
1.86 ~ 1.68 ( m, 8H, CCH2C )。 ESI-M S m /z:
503.55 [M + H]+ 。
0155g ( 8016% ), m p 129 ~ 131 e 。
1H-NM R
( CDC l3 ) D: 7.28( s, 4H, A r-H ), 3.55 ( br s, 4H,A r-CH2 ), 2.82 ~ 2.52( m, 32H, NCH2, NHCH2 ),
1.86 ~ 1.68 ( m, 8H, CCH2C )。 ESI-M S m /z:
503.55 [M + H]+ 。
...............................................
SEE
.........................................

...............................
U.S. Pat. No. 5,021,409 is directed to a method of treating retroviral infections comprising administering to a mammal in need of such treatment a therapeutically effective amount of a bicyclic macrocyclic polyamine compound. Although the usefulness of certain alkylene and arylene bridged cyclam dimers is generically embraced by the teachings of the reference, no arylene bridged cyclam dimers are specifically disclosed.
WO 93/12096 discloses the usefulness of certain linked cyclic polyamines in combating HIV and pharmaceutical compositions useful therefor. Among the specifically disclosed compounds is 1,1'- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11 tetraazacyclotetradecane (and its acid addition salts), which compound is a highly potent inhibitor of several strains of human immune deficiency virus type 1 (HIV-1) and type 2 (HIV-2).
European Patent Appln. 374,929 discloses a process for preparing mono-N-alkylated polyazamacrocycles comprising reacting the unprotected macrocycle with an electrophile in a non-polar, relatively aprotic solvent in the absence of base. Although it is indicated that the monosubstituted macrocycle is formed preferentially, there is no specific disclosure which indicates that linked bicyclams can be synthesized by this process.
U.S. Pat. No. 5,047,527 is directed to a process for preparing a monofunctionalized (e.g., monoalkylated)cyclic tetramine comprising: 1) reacting the unprotected macrocycle with chrominum hexacarbonyl to obtain a triprotected tetraazacyloalkane compound; 2) reacting the free amine group of the triprotected compound prepared in 1) with an organic (e.g., alkyl) halide to obtain a triprotected monofunctionalized (e.g., monoalkylated) tetraazacycloalkane compound; and 3) de-protecting the compound prepared in 2) by simple air oxidation at acid pH to obtain the desired compound. In addition, the reference discloses alternative methods of triprotection employing boron and phosphorous derivatives and the preparation of linked compounds, including the cyclam dimer 1,1'- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane, by reacting triprotected cyclam prepared as set forth in 1) above with an organic dihalide in a molar ratio of 2:1, and deprotecting the resultant compound to obtain the desired cyclam dimer.
J. Med. Chem., Vol. 38, No. 2, pgs. 366-378 (1995) is directed to the synthesis and anti-HIV activity of a series of novel phenylenebis(methylene)-linked bis-tetraazamacrocyclic analogs, including the known cyclam dimer 1,1'- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane. The cyclam dimers disclosed in this reference, including the afore-mentioned cyclam dimer, are prepared by: 1) forming the tritosylate of the tetraazamacrocycle; 2) reacting the protected tetraazamacrocycle with an organic dihalide, e.g., dibromo-p-xylene, in acetonitrile in the presence of a base such as potassium carbonate; and 3) de-protecting the bis-tetraazamacrocycle prepared in 2) employing freshly prepared sodium amalgam, concentrated sulfuric acid or an acetic acid/hydrobromic acid mixture to obtain the desired cyclam dimer, or an acid addition salt thereof.
is directed to the synthesis and anti-HIV activity of a series of novel phenylenebis(methylene)-linked bis-tetraazamacrocyclic analogs, including the known cyclam dimer 1,1'- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane. The cyclam dimers disclosed in this reference, including the afore-mentioned cyclam dimer, are prepared by: 1) forming the tritosylate of the tetraazamacrocycle; 2) reacting the protected tetraazamacrocycle with an organic dihalide, e.g., dibromo-p-xylene, in acetonitrile in the presence of a base such as potassium carbonate; and 3) de-protecting the bis-tetraazamacrocycle prepared in 2) employing freshly prepared sodium amalgam, concentrated sulfuric acid or an acetic acid/hydrobromic acid mixture to obtain the desired cyclam dimer, or an acid addition salt thereof.
Although the processes disclosed in U.S. Pat. No. 5,047,527 and the J. Med. Chem. reference are suitable to prepare the cyclam dimer 1,1'- 1,4-phenylene bis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane, they involve the use of cyclam as a starting material, a compound which is expensive and not readily available. Accordingly, in view of its potent anti-HIV activity, a number of research endeavors have been undertaken in an attempt to develop a more practical process for preparing 1,1'- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane.
are suitable to prepare the cyclam dimer 1,1'- 1,4-phenylene bis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane, they involve the use of cyclam as a starting material, a compound which is expensive and not readily available. Accordingly, in view of its potent anti-HIV activity, a number of research endeavors have been undertaken in an attempt to develop a more practical process for preparing 1,1'- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane.
EXAMPLE 1
a) Preparation of the 1,4-phenylenebis-methylene bridged hexatosyl acylic precursor of formula III
To a 4-necked, round-bottom flask, equipped with a mechanical stirrer, heating mantle, internal thermometer and addition funnel, is added 43.5 g (0.25 mol) of N,N'-bis(3-aminopropyl) ethylenediamine and 250 ml of tetrahydrofuran. To the resultant solution is added, over a period of 30 minutes with external cooling to maintain the temperature at 20° C., 113.6 g (0.8 mol) of ethyl trifluoroacetate. The reaction mixture is then stirred at room temperature for 4 hours, after which time 52.25 ml. (0.3 mol) of diisopropylethylamine is added. The resultant reaction mixture is warmed to 60° C. and, over a period of 2 hours, is added a solution of 33.0 g (0.125 mol) of α,α'-dibromoxylene in 500 ml. of tetrahydrofuran. The reaction mixture is then maintained at a temperature of 60° C., with stirring, for an additional 2 hours after which time a solution of 62.0 g. (1.55 mol) of sodium hydroxide in 250 ml. of water is added. The resultant mixture is then stirred vigorously for 2 hours, while the temperature is maintained at 60° C. A solution of 152.5 g. (0.8 mol) of p-toluenesulfonyl-chloride in 250 ml. of tetrahydrofuran is then added, over a period of 30 minutes, while the temperature is maintained at between 20° C. and 30° C. The reaction is then allowed to proceed for another hour at room temperature. To the reaction mixture is then added 1 liter of isopropyl acetate, the layers are separated and the organic layer is concentrated to dryness under vacuum to yield the desired compound as a foamy material.
b) Preparation of the hexatosyl cyclam dimer of formula IV
To a 4-necked, round-bottom flask, equipped with a mechanical stirrer, heating mantle, internal thermometer and addition funnel, is added 114.6 g. (0.10 mol) of the compound prepared in a) above and 2.5 liters of dimethylformamide. After the system is degassed, 22.4 g. (0.56 mol) of NaOH beads, 27.6 g (0.2 mol) of anhydrous potassium carbonate and 5.43 g. (0.016 mol) of t-butylammonium sulfate are added to the solution, and the resultant mixture is heated to 100° C. and maintained at this temperature for 2.5 hours. A solution of 111.0 g (0.3 mol) of ethyleneglycol ditosylate in 1 liter of dimethylformamide is then added, over a period of 2 hours, while the temperature is maintained at 100° C. After cooling the reaction mixture to room temperature, it is poured into 4 liters of water with stirring. The suspension is then filtered and the filter cake is washed with 1 liter of water. The filter cake is then thoroughly mixed with 1 liter of water and 2 liters of ethyl acetate. The solvent is then removed from the ethyl acetate solution and the residue is re-dissolved in 500 ml. of warm acetonitrile. The precipitate that forms on standing is collected by filtration and then dried to yield the desired compound as a white solid.
c) Preparation of 1,1'- 1,4-phenylenebis-(methylene)!-bis-1,4,8,11-tetraazacyclotetradecane
In a 4-necked, round-bottom flask, equipped with a mechanical stirrer, heating mantle, internal thermometer and addition funnel, is added 26.7 g.(0.02 mol) of the compound prepared in b) above, 300 ml. of 48% hydrobromic acid and 1 liter of glacial acetic acid. The resultant mixture is then heated to reflux and maintained at reflux temperature, with stirring, for 42 hours. The reaction mixture is then cooled to between 22° C. and 23° C. over a period of 4 hours, after which time it is stirred for an additional 12 hours. The solids are then collected using suction filtration and added to 400 ml. of deionized water. The resultant solution is then stirred for 25 to 30 minutes at a temperature between 22° C. and 23° C. and filtered using suction filtration. After washing the filter pad with a small amount of deionized water, the solution is cooled to between 10° C. and 15° C. 250 g. of a 50% aqueous solution of sodium hydroxide is then added, over a period of 30 minutes, while the temperature is maintained at between 5° C. and 15° C. The resultant suspension is stirred for 10 to 15 minutes, while the temperature is maintained at between 10° C. and 15° C. The suspension is then warmed to between 22° C. and 23° C. and to the warmed suspension is added 1.5 liters of dichloromethane. The mixture is then stirred for 30 minutes, the layers are separated and the organic layer is slurried with 125 g. of sodium sulfate for 1 hour. The solution is then filtered using suction filtration, and the filtrate is concentrated under reduced pressure (40°-45° C. bath temperature, 70-75 mm Hg) until approximately 1.25 liters of solvent is collected. To the slurry is then added 1.25 liters of acetone, and the filtrate is concentrated under reduced pressure (40°-45° C. bath temperature, 70-75 mm Hg) until approximately 1.25 liters of solvent is collected. The slurry is then cooled to between 22° C. and 23° C. and the solids are collected using suction filtration. The solids are then washed with three 50 ml. portions of acetone and dried in a vacuum oven to obtain the desired compound as a white solid.
EXAMPLE 2
The following is an alternate procedure for the preparation of the 1,4-phenylenebis-methylene bridged hexatosyl acyclic precursor of formula III.
To a 3-necked, round-bottomed flask, equipped with a mechanical stirrer, heating mantle, internal thermometer and addition funnel, is added 3.48 g. (20 mmol) of N,N'-bis-(3-aminopropyl)ethylenediamine and 20 ml. of tetrahydrofuran. To the resultant solution is added, over a period of 20 minutes with external cooling to maintain the temperature at 20° C., 5.2 ml. (42 mmol) of ethyl trifluoroacetate. The reaction mixture is then stirred at room temperature for 1 hour, after which time a solution of 2.64 g. (10 mmol) of α,α'-dibromoxylene in 20 ml. of tetrahydrofuran is added. The resultant reaction mixture is then stirred at room temperature for 4 hours. A solution of 4.8 g. (120 mmol) of sodium hydroxide in 20 ml. of water is then added and the resultant mixture is warmed to 60° C. and maintained at this temperature, with vigorous stirring, for 2 hours. Over a period of 20 minutes, 13.9 g. (73 mmol) of p-toluenesulfonylchloride is then added portionwise, while the temperature is maintained at 20° C. The reaction is then allowed to proceed for another hour at room temperature. To the reaction mixture is then added 100 ml. of isopropyl acetate, the layers are separated and the organic layer is washed with saturated sodium bicarbonate aqueous solution. The solution is then condensed to 40 ml., cooled to 4° C. and kept at that temperature overnight. The resultant suspension is filtered and the solid is washed with 10 ml. of isopropyl acetate. The solvents are then removed from the filtrate to yield the desired compound as a brown gel.
..............................
see
Synthesis and structure-activity relationships of phenylenebis(methylene)linked bis-tetraazamacrocycles that inhibit HIV replication. Effects of macrocyclic ring size and substituents on the aromatic linker
J Med Chem 1995, 38(2): 366
J Med Chem 1995, 38(2): 366
.........................................................
see
New bicyclam-AZT conjugates: Design, synthesis, anti-HIV evaluation, and their interaction with CXCR-4 coreceptor
J Med Chem 1999, 42(2): 229
J Med Chem 1999, 42(2): 229
..............................................................
CN 102584732
[0003]

[0004] plerixafor (trade name Mozobil ™) was developed by the U.S. company Genzyme chemokine receptor 4 (CXCR4) antagonist specificity. The drug is a hematopoietic stem (progenitor) cell activator, and can stimulate hematopoietic stem cell proliferation and differentiation into functional blood circulation.
[0005] As the non-Hodgkin's lymphoma (NHL) and multiple myeloma (Korea) most of the cases and the progress of cases to alleviate the need for autologous peripheral blood stem cell transplantation, and plerixafor joint G-CSF can significantly improve the number of patients with ⑶ 34 + cells, about 60% of the patient's peripheral blood can ⑶ 34 + cells increased to ensure that the NHL and MM patients with autologous hematopoietic stem cell transplantation success.
[0006] U.S. FDA approval on December 15, 2008 its listing, clinical studies showed that the drug can greatly increase the number of white blood cells of patients and to promote hematopoietic stem cells from bone marrow to the blood flow, and granulocyte colony-stimulating factor (G-CSF ) have a synergistic effect; has been used in multiple myeloma and Hodgkin's lymphoma patients with stem cell transplantation in clinical trials.
[0007] About plerixafor or synthetic analogs have some at home and abroad reported in the literature, there are J.0rg.Chem.2003, 68,6435-6436; J.Med Chem.1995, 38 (2): 366-378; J.SynthCommun.1998 ,28:2903-2906; Tetrahedron, 1989,45 (1) :219-226; Chinese Journal of Pharmaceuticals 2007,38 (6); World Patent W09634860A1; W09312096A1; U.S. Patent US5047527, US5606053, US5801281, US5064956, Chinese patent CN1466579A.
[0008] J.Med Chem.1995, 38 (2) = 366-378 relates to a preparation method comprises the following steps: a) forming a salt of trimethoxy benzene tetraaza macrocycles; 2) reacting the protected tetrazole hetero macrocycle in acetonitrile under the presence of a base such as potassium carbonate as dibromo-p-xylene is reacted with an organic dihalide; 3) using freshly prepared sodium amalgam, concentrated sulfuric acid or acetic acid / hydrobromic acid mixture deprotected target product.
[0009] US 5047527 relates to preparation of the cyclic four monofunctional amine, the method comprising: a) reacting the unprotected macrocycle of reaction with chromium hexacarbonyl to obtain protection tetraazadecalin three compounds; 2) 3 Protection of the free amino compound with an organic halide to obtain three-protected monofunctional tetraaza naphthenic compounds; 3) simple air oxidation, deprotection to obtain the desired product. [0010] J.Synth Commun.1998 ,28:2903-2906 describes an improved method for synthesizing intermediates Plerixafor, the method using phosphor protection, deprotection to give a smooth 1,1 '- [1,4 - phenylene bis (methylene)] _ two _1, 4,8,11 - tetraazacyclododecane fourteen burn.
[0011] US 5606053 relates to a process for preparing dimers 1, I '- [1,4 - phenylene bis (methylene)] - two -1,4,8,11 - tetraazacyclododecane-tetradecane method. The preparation of compounds include: 1) the four-amine as the starting material, obtained by acylation of toluene Juan acyclic intermediates and three xylene sulfonate and toluene sulfonate and toluene intermediates; 2) and xylene sulfonate and intermediates trimethylbenzene toluenesulfonic acid intermediates after alkylation separation dibromo xylene, toluene sulfonate and then obtain a non-cyclic dimers of six toluenesulfonic acylated; 3) six isolated bridged acyclic toluenesulfonic acid dimer form is reacted with ethylene glycol ditosylate three equivalents of cyclization; 4) deprotection to obtain the objective product was purified by hydrobromic acid and acetic acid.
[0012] US 5801281 relates to preparation of dimer 1, I '- [1,4 _-phenylene bis (methylene)] - two _1, 4,8,11
[0013] - tetraazacyclo tetradecane, comprising: a) reacting the acyclic tetraamine with 3 equivalents of ethyl trifluoroacetate, the reaction; 2) with 0.5 equivalents of the tri-dibromo-p-xylene-protected acyclic alkylation of the amine obtained form four non-cyclic dimers; 3) hydrolysis to remove the six trifluoroacetyl compound group; 4) acylation of the compound toluenesulfonic bridged tetraamine dimer; 5) B Juan xylene glycol ester cyclization; 6) and glacial acetic acid mixed with hydrobromic acid deprotection was the target product.
Under the [0014] US 5064956 discloses a multi-alkylated single-ring nitrogen of the compound prepared, the method involves reacting the unprotected macrocycle in an aprotic, relatively non-polar solvent in presence of alkali electrophilic reagent. Not mentioned in this document similar to the embodiment Seclin dimer synthesis.
[0015] Through the open Plerixafor synthetic route research and meta-analysis of the literature, mainly in the following four synthetic routes:
[0016] Route One, is 1,4,8,11 - tetraazacyclododecane cyclotetradecane as raw material, NI, N4, N8 three protected with 1,4 - bis (halomethyl) benzene-bridged deprotection to obtain the finished product. The following reaction scheme, wherein R is p-toluenesulfonyl group, a methanesulfonyl group, a trifluoroacetyl group, a tert-butoxycarbonyl group and the like:
[0017]

[0018] Route II is di (2 - aminopropyl) ethylenediamine as raw material, the ring and the reaction with 1,4 - bis (halomethyl) benzene-bridged, and then deprotection Bullock Suffolk.
[0019] Route 3 to 1,4,8,11 - tetraazacyclododecane cyclotetradecane as raw material, under anhydrous, anaerobic conditions, after the ring protection with 1,4 - bis (halomethyl ) benzene bridging, and then deprotection plerixafor. Synthesis scheme below, wherein R is P, Ni, etc.;

[0021] line four, based on acrylate as starting material, first with ethylene diamine as raw material by Michael addition of the amine solution, then with malonate cyclization 1,4,8,11 - Tetraaza _5, 7,12 - three oxo cyclotetradecane by α, α '- dibromo-p-xylene bridging, the final deprotection plerixafor. Reaction Roadmap follows:
[0022]

[0023] The above synthesis route and the existing methods have the following disadvantages:
[0024] In an intermediate of the synthesis route, the existing technology, the need for column purification of the intermediates, low yield.
[0025] route to protect the stability of the two because of the strong, leading to the final deprotection step difficult, long production cycle, low yield, and finished organic residues can not be achieved within the standard limits.
Higher dry anaerobic demands [0026] Route 3 on, harsh reaction conditions, deprotection is not complete, intermediates need to repeatedly purified, low yield, after repeated recrystallization, finished monohetero difficult to control in 0.1% less.
[0027] Anhydrous ethylene diamine route and need four anhydrous THF, more stringent requirements on the process, and to use dangerous borane dimethyl sulfide, while the second step is only about 35% lower yield. Selectivity of the reaction is not high shortcomings, so do not be the most economical and reasonable synthetic route.
[0028] We prepared by Plerixafor prepared by methods disclosed above may Plerixafor single impurity of 0.1% or less is difficult to achieve, it is difficult to meet the quality requirements of the injection material, the same techniques can not reach the European Quality of ICH guidelines of the relevant technical requirements, low yield, high cost required for each step of the intermediate column to afford a large amount of solvent, time consuming, and the greater the elution solvent toxicity, is not suitable for industrial production.
(I) Preparation of 1,4,8 _ tris (p-toluenesulfonyl) -1,4,8,11 - tetraazacyclododecane-tetradecane: the raw 1,4,8,11 - tetraazacyclododecane cyclotetradecane suspended in methylene chloride, in the role of acid binding agent, at a temperature 10 ~ 30 ° C, p-toluenesulfonyl chloride and 3 ~ 8h, filtered, and the filtrate was collected and concentrated to dryness to obtain a residue; will have The residue of said C ^ C3 alkyl group in a mixed solvent of alcohol and an aprotic solvent, purification, crystallization segment greater than 95% purity of 1,4,8 - tris (p-toluenesulfonyl) _1, 4,8,11 - tetraaza cyclotetradecane;
[0032] (2) Preparation of 1,1 '- [1,4 - (phenylene methylene)] - two - [4,8,11 - tris (p-toluenesulfonyl)] -1,4, 8,11 - tetraazacyclododecane-tetradecane: A (I) the resulting 1,4,8 - tris (p-toluenesulfonyl) _1, 4,8,11 - tetraazacyclododecane-tetradecane, α, α two bromo-p-xylene in place of anhydrous acetonitrile, was added acid-binding agent, the reaction was refluxed under nitrogen for 5 to 24 hours; After the reaction was cooled to room temperature, the reaction mixture was then collected by filtration and the filter cake was purified to obtain a mixed solvent I , I, - [1,4 - (phenylene methylene)] - two - [4,8,11 - tris (p-toluenesulfonyl)] _1, 4,8,11 - tetraazacyclododecane ten four alkyl;
[0033] (3) Synthesis Plerixafor: A (2) the resultant I, 1'-[1,4 _ (phenylene methylene)] - two - [4,8,11 - tris (p-toluene sulfonyl)] -1,4,8,11 - tetraazacyclododecane myristic acid solution was added to the mixture, stirred and dissolved, the reaction was warmed to reflux for 10 to 24 hours, cooled, filtered, and filter cake was collected; the filter cake was dissolved in purified water, adjusted with sodium hydroxide solution or potassium hydroxide solution to the PH-12, filtered, and the filtrate was extracted with a halogenated solvent, and the organic layer was dried over anhydrous sodium sulfate and then filtered, the filtrate was concentrated under reduced pressure P Le Suffolk crude;
[0034] (4) Purification Plerixafor: Plerixafor the crude was dissolved into a solvent and heated to reflux to dissolve, filtered, and the crystallization solvent is added dropwise at 40 ~ 45 ° C crystallization 30min, filtered and the filtrate then cooled to 20 ~ 25 ° C crystallization I hour at O ~ 5 ° C crystallization three hours, filtered, and the filter cake was dried Plerixafor.
Plerixafor Preparation: 6 [0075] Implementation
[0076] The starting material 1,4,8,11 - tetraazacyclo tetradecane (5g, 25mmol) was suspended in dichloromethane (50g) was added N, N-diisopropylethylamine (7.5ml) , a solution of p-toluenesulfonyl chloride (10.8g, 56.5mmol) and methylene chloride (50g) in a solution of, at 25 ~ 30 ° C reaction temperature 3h, filtered, and the filtrate was collected and concentrated to dryness and to the residue in methanol (30g), toluene (IOg) was heated to reflux, filtered, and the filtrate was cooled to 40 ° C crystallization 30min, filtered to remove impurities little over protection, and the filtrate was added methyl tert-butyl ether (30g), stirring rapidly cooled to O ~ 5 ° C crystallization 3h, filtered, and dried to give 1,4,8 - tris (p-toluenesulfonyl) -1, 4,8,11 - tetraazacyclododecane-tetradecane (9.6g, 61.9%), purity of 97.2%.
[0077] The 4,8 _ tris (p-toluenesulfonyl) _1, 4,8,11 - tetraazacyclododecane-tetradecane (9g, 13.6mmol) α, α '- dibromo-p-xylene (1.81 g, 6.8mmol) in dry acetonitrile was placed (90ml) was added potassium carbonate (15.0g, 108.5mmol), the reaction was refluxed under nitrogen for 5 hours. Cooled to room temperature and filtered to collect the filter cake, was added anhydrous methanol (10ml), ethyl acetate (30ml), dichloromethane (IOml) hot melt, whereby the cooling crystallization, filtration, and dried under reduced pressure to obtain white solid (16. lg, 83%), purity 97.5%.
[0078] The intermediate obtained above (5g, 3.5mmol) was added to glacial acetic acid (25ml) and concentrated hydrochloric acid (25ml) was stirred until dissolved in the mixed solution was heated to reflux for 24 hours, cooled, collected by filtration cake. The filter cake was dissolved in purified water (20ml), adjusting the PH value of the solution with sodium hydroxide to 12, filtered, and the filtrate was extracted with dichloromethane (50mlX3), the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain sand Bullock Fu crude (1.4g, 79.5%), purity 98.6%.
[0079] The crude Plerixafor (1.4g) is placed in tetrahydrofuran (14g), heated to reflux to dissolve, filtered, and added dropwise n-hexane (42g), and 40 ~ 45 ° C crystallization 30min, filtered little solid, The filtrate was rapidly cooled to 20 ~ 25 ° C crystallization I hour and then at O ~ 5 ° C crystallization three hours, filtered, 45 ° C and dried under reduced pressure to obtain the finished Plerixafor (1.2g, 85.7%), purity 99.93 %, the largest single miscellaneous 0.04%.
........................................

wherein, n is 0 or 1, Ts is tosyl radical, P is trifluoroacetyl or p-tosyl radical;
To the NaOH solution of the starting material 7 is dropwise added ether solution of tosyl chloride. The system is stirred over night. A white solid is formed and filtrated. The filter cake is washed with water and ethyl ether, respectively, recrystallized to give a white solid intermediate of formula 8. To the dried acetonitrile solution of the compound of formula 8 is slowly dropwise added dried acetonitrile solution of 1,2-di-p-tosyloxypropane under reflux state, refluxed for 2-4 days, stood until room temperature. A white solid is precipitated and filtrated. The filter cake is washed with water and ethyl acetate, respectively, recrystallized to give a white solid compound of formula 9. The compound of formula 9 is dissolved in 90% concentrated sulfuric acid, allowed to react at 100° C. for 24-48 hours, stood until room temperature. To the reaction solution are dropwise added successively ethanol and ethyl ether. A white solid is precipitated, filtrated, dried, and dissolved in NaOH solution. The aqueous phase is extracted with chloroform. The chloroform phase is combined, concentrated, recrystallized to give a white solid compound of formula 10. To the chloroform solution of the compound of formula 10 and triethylamine is dropwise added chloroform solution of tosyl chloride. The mixture is allowed to react at room temperature over night, concentrated and column separated (eluant: dichloromethane/methanol system) to give a white solid compound of formula 11 (protective group is tosyl); or to the methanol solution of the compound of formula 10 is dropwise added ethyl trifluoroacetate. The mixture is allowed to react at room temperature over night, concentrated and column separated (eluant: ethyl acetate) to give a white solid compound of formula 11 (protective group is trifluoroacetyl);
To the NaOH solution of the starting material 7 is dropwise added ether solution of tosyl chloride. The system is stirred over night. A white solid is formed and filtrated. The filter cake is washed with water and ethyl ether, respectively, recrystallized to give a white solid intermediate of formula 8. To the dried acetonitrile solution of the compound of formula 8 is slowly dropwise added dried acetonitrile solution of 1,2-di-p-tosyloxypropane under reflux state, refluxed for 2-4 days, stood until room temperature. A white solid is precipitated and filtrated. The filter cake is washed with water and ethyl acetate, respectively, recrystallized to give a white solid compound of formula 9. The compound of formula 9 is dissolved in 90% concentrated sulfuric acid, allowed to react at 100° C. for 24-48 hours, stood until room temperature. To the reaction solution are dropwise added successively ethanol and ethyl ether. A white solid is precipitated, filtrated, dried, and dissolved in NaOH solution. The aqueous phase is extracted with chloroform. The chloroform phase is combined, concentrated, recrystallized to give a white solid compound of formula 10. To the chloroform solution of the compound of formula 10 and triethylamine is dropwise added chloroform solution of tosyl chloride. The mixture is allowed to react at room temperature over night, concentrated and column separated (eluant: dichloromethane/methanol system) to give a white solid compound of formula 11 (protective group is tosyl); or to the methanol solution of the compound of formula 10 is dropwise added ethyl trifluoroacetate. The mixture is allowed to react at room temperature over night, concentrated and column separated (eluant: ethyl acetate) to give a white solid compound of formula 11 (protective group is trifluoroacetyl);
Pharmacokinetics
Following subcutaneous injection, plerixafor is absorbed quickly and peak concentrations are reached after 30 to 60 minutes. Up to 58% are bound to plasma proteins, the rest mostly resides in extravascular compartments. The drug is not metabolized in significant amounts; no interaction with the cytochrome P450 enzymes or P-glycoproteins has been found. Plasma half life is 3 to 5 hours. Plerixafor is excreted via the kidneys, with 70% of the drug being excreted within 24 hours.[5]
Pharmacodynamics
In the form of its zinc complex, plerixafor acts as an antagonist (or perhaps more accurately a partial agonist) of the alpha chemokine receptor CXCR4 and an allosteric agonist ofCXCR7.[10] The CXCR4 alpha-chemokine receptor and one of its ligands, SDF-1, are important in hematopoietic stem cell homing to the bone marrow and in hematopoietic stem cell quiescence. The in vivo effect of plerixafor with regard to ubiquitin, the alternative endogenous ligand of CXCR4, is unknown. Plerixafor has been found to be a strong inducer of mobilization of hematopoietic stem cells from the bone marrow to the bloodstream as peripheral blood stem cells.[11]
Interactions
No interaction studies have been conducted. The fact that plerixafor does not interact with the cytochrome system indicates a low potential for interactions with other drugs.[5]
Legal status
Plerixafor has orphan drug status in the United States and European Union for the mobilization of hematopoietic stem cells. It was approved by the U.S. Food and Drug Administration for this indication on December 15, 2008.[12] In Europe, the drug was approved after a positive Committee for Medicinal Products for Human Use assessment report on 29 May 2009.[7] The drug was approved for use in Canada by Health Canada on December 8, 2011.[13]
Research
Small molecule cancer therapy
Plerixafor was seen to reduce metastasis in mice in several studies.[14] It has also been shown to reduce recurrence of glioblastoma in a mouse model after radiotherapy. In this model, the cancer surviving radiation are critically depended on bone marrow derived cells for vasculogenesis whose recruitment mediated by SDF-1 CXCR4 interaction is blocked by plerixafor.[15]
Use in generation of other stem cells
Researchers at Imperial College have demonstrated that plerixafor in combination with vascular endothelial growth factor (VEGF) can produce mesenchymal stem cells andendothelial progenitor cells in mice.[16]
Other uses
Blockade of CXCR4 signalling by plerixafor (AMD3100) has also unexpectedly been found to be effective at counteracting opioid-induced hyperalgesia produced by chronic treatment with morphine, though only animal studies have been conducted as yet.[17]
 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
| 1,1′-[1,4-Phenylenebis(methylene)]bis [1,4,8,11-tetraazacyclotetradecane] | |
| Clinical data | |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a609018 |
| Pregnancy cat. | D (US) |
| Legal status | ℞-only (US) |
| Routes | Subcutaneous injection |
| Pharmacokinetic data | |
| Protein binding | Up to 58% |
| Metabolism | None |
| Half-life | 3–5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS number | 110078-46-1 |
| ATC code | L03AX16 |
| PubChem | CID 65015 |
| IUPHAR ligand | 844 |
| DrugBank | DB06809 |
| ChemSpider | 58531 |
| UNII | S915P5499N |
| Synonyms | JM 3100, AMD3100 |
| Chemical data | |
| Formula | C28H54N8 |
| Mol. mass | 502.782 g/mol |
(Plerixafor), chemical name: 1, I '- [I, 4_ phenylene ni (methylene)] - ni -1,4,
8,11 - tetraazacyclo tetradecane, its molecular structure is as follows:
[0004]

Synthesis of domestic and foreign literature in general, all require 1,4,8,11 - tetraazacyclo-tetradecane for 3 protection (eg of formula I), of the three methods are used to protect the p-toluenesulfonamide chloride, trifluoroacetic acid ko ko cool, tert-butyl carbonate ni. Use of p-toluenesulfonamide-protected deprotection step into strict step because deprotecting reagent (such as hydrobromic acid / glacial acetic acid, concentrated sulfuric acid, etc.) side reactions often occur.The use of trifluoroacetic acid ko ko ester protecting, since the trifluoromethyl group strongly polar ko, resulting fourth-NH unprotected decrease in activity, usually not fully reflect the subsequent reaction, thereby further into ー is introduced after deprotection difficult to remove impurities 1,4,8,11 - tetraazacyclo-tetradecane.
[0006] tert-butyl carbonate ni selective protection of the amino group is widely used (polyamines, amino acids, p printed tidic chains, etc.), but to use it for 1,4,8,11 - tetraazacyclo tetradecane rarely reported, abroad it for 1,4,8,11 - tetraazacyclo tetradecane protection coverage, we use the t-butyl carbonate brother attempted 3 protection, he was surprised to find that in certain conditions, the three protection up to 90% (see Figure I), with high selectivity, significantly higher than the reported domestic Boc protected
Selectivity of the reaction (see table below).
[0007]

[0008] 2 by three protection product with quite different polarity protection products, flash column chromatography using silica gel column to separate the protector 3 of sufficient purity, and deprotection conditions milder (only hydrochloric acid solution), in a certain extent reduce the incidence of side effects, so capable of synthesizing high purity products.
[0009]

SUMMARY OF THE INVENTION


xample I: 3Boc protection 1,4,8,11 _ tetraazacyclo Preparation tetradecane
[0048] 1,4,8,11 taken tetraazacyclo tetradecane _ 10g (0.05mol), and acetone - water (2: l) 50ml, tris ko amine 10. 119g (0. Lmol), ni ko isopropyl amine 3. 225g (0. 025mol), at room temperature was added dropwise tert-butyl carbonate, brother 38. 194g (0. 175mol), dropwise at room temperature after stirring for 24 hours, HPLC monitoring of the reaction. After completion of the reaction 50 ° C under reduced pressure to dryness to give a pale yellow oil, 150g on a silica gel column, and eluted with ko acid esters ko collecting ko ko acid ester liquid evaporated to dryness under reduced pressure to give a white foam 23. 12g, yield of 92.36%. 1HNMR (400MHz, CDCl3, 6 ppm): 1. 74 (2H, q, 5. 5);
I. 96 (2H, q, 6. 5); 2. 66 (2H, t, 5. 5); 2. 82 (2H, t, 5. 5); 3. 33 (4H, m); 3. 34 (2H, m); 3. 37 (2H, m), 3. 43 (4H, m).
[0049] Implementation Example 2: 6Boc protection Bullock Suffolk Preparation
[0050] Take 3Boc protection 1,4,8,11 _ tetraazacyclo tetradecane 20. 03g (0. 04mol), dissolved in anhydrous ko nitrile 400ml, anhydrous potassium carbonate 20g, aa 'ni chlorine ni toluene 3.5012g (0.02mol), sodium iodide 75mg, at reflux for 24 hours under nitrogen, TLC monitoring of the reaction. After completion of the reaction, cooled to room temperature, filtered, the filter cake was washed with 200ml of ko nitrile, nitrile ko combined solution was evaporated to dryness under reduced pressure to give the protected Bullock 6Boc Suffolk 21. 20g, yield of 96.06%. Alcohol with ko - a mixed solvent of water and recrystallized to give a white solid. [0051] Implementation Example 3: Bullock Suffolk • 8HC1 • 3H20 Preparation of compounds
[0052] Protection Bullock Suffolk take 6Boc 20g, add methanol 200ml, stirring to dissolve, concentrated hydrochloric acid was added dropwise at room temperature, 60ml, was stirred at room temperature after the addition was complete 48 inches, TLC monitoring of the reaction. After completion of the reaction, filtration, the filter cake was dried 50 ° C under reduced pressure to give a white solid 13. 54g, yield of 88.04%.

[0053] Implementation Example 4: Preparation of Suffolk Bullock............Plerixafor BASE
[0054] Take Bullock Suffolk • 8HC1 • 3H20 compound 13. 54g, add water 40ml ultrasound to dissolve after stirring constantly with 50% sodium hydroxide solution to adjust the pH to 12 and filtered, the filter cake 50 ° C minus pressure and dried to give a white solid 7. 24g, yield 90.24 V0o
1H NMR (400MHz, CDCl3, 6 ppm): 1. 75 (4H, bs); 1. 87 (4H, bs); 2. 95-2. 51 (32H, m); 3. 54 (4H, s); 4. 23 (4H, bs); 7. 30 (4H, s).
IR (KBr) 3280,2927,2883,2805,1458,1264,1117 cm,
NEW PATENT................WO-2014125499
Improved and commercially viable process for the preparation of high pure plerixafor base
Process for the preparation of more than 99.8% pure plerixafor base by HPLC. Also claims solid forms of plerixafor base and composition comprising the same. Appears to be the first filing from the assignee on this API. FDA Orange book lists US6987102 and US7897590, expire in July 2023.
3-5-1997
|
Process for preparing 1,4,8,11-tetraazacyclotetradecane
| |
2-26-1997
|
Process for preparing 1,1'-[1,4-phenylenebis-(methylene)]-bis-1,4,8,11-tetraazacyclotetradecane
| |
12-11-1996
|
Aromatic-linked polyamine macrocyclic compounds with anti-HIV activity
| |
11-8-1996
|
PROCESS FOR PREPARING 1,1'-[1,4-PHENYLENEBIS-(METHYLENE)]-BIS-1,4,8,11-TETRAAZACYCLOTETRADECANE
| |
10-4-1996
|
PROCESS FOR PREPARING 1,1'-[1,4-PHENYLENEBIS-(METHYLENE)]-BIS-1,4,8,11-TETRAAZACYCLOTETRADECANE
| |
7-14-1995
|
CYCLIC POLYAMINES
| |
6-25-1993
|
LINKED CYCLIC POLYAMINES WITH ACTIVITY AGAINST HIV
|
9-2-2005
|
Substituted benzodiazepines as inhibitors of the chemokine receptor CXCR4
| |
2-4-2005
|
Methods and compositions for the treatment or prevention of human immunodeficiency virus and related conditions using cyclooxygenase-2 selective inhibitors and antiviral agents
| |
12-4-2002
|
Process for preparation of N-1 protected N ring nitrogen containing cyclic polyamines and products thereof
| |
10-2-2002
|
Prodrugs
| |
10-25-2001
|
PROCESS FOR PREPARING 1,1'- 1,4-PHENYLENEBIS-(METHYLENE)]-BIS-1,4,8,11-TETRAAZACYCLOTETRADECANE
| |
9-29-2000
|
CHEMOKINE RECPETOR BINDING HETEROCYCLIC COMPOUNDS
| |
8-11-2000
|
METHODS AND COMPOSITIONS TO ENHANCE WHITE BLOOD CELL COUNT
| |
1-15-1998
|
PROCESS FOR PREPARING 1,1'- 1,4-PHENYLENEBIS-(METHYLENE) -BIS-1,4,8,11-TETRAAZACYCLOTETRADECANE
| |
3-19-1997
|
Process for preparing 1,1'-[1,4-phenylenebis-(methylene)]-bis-1,4,8,11-tetraazacyclotetradecane
| |
3-7-1997
|
PROCESS FOR PREPARING 1,4,8,11-TETRAAZACYCLOTETRADECANE PROCESS FOR PREPARING 1,4,8,11-TETRAAZACYCLOTETRADECANE
|
6-24-2011
|
BETULINIC ACID DERIVATIVES AS ANTI-HIV AGENTS
| |
11-3-2010
|
Antiviral methods employing double esters of 2', 3'-dideoxy-3'-fluoroguanosine
| |
2-5-2010
|
Chemokine Receptor Modulators
| |
1-29-2010
|
NOVEL POLYNITROGENATED SYSTEMS AS ANTI-HIV AGENTS
| |
9-4-2009
|
Combination of CXCR4 Antagonist and Morphogen to Increase Angiogenesis
| |
11-28-2008
|
Chemokine receptor modulators
| |
10-24-2008
|
Chemokine receptor modulators
| |
8-32-2006
|
Compositions and methods for treating tissue ischemia
| |
7-5-2006
|
ANTIVIRAL METHODS EMPLOYING DOUBLE ESTERS OF 2', 3'-DIDEOXY-3'-FLUOROGUANOSINE
| |
12-14-2005
|
Treatment of viral infections using prodrugs of 2',3-dideoxy,3'-fluoroguanosine
|
References
- Jump up^ Ciampolini, M.; Fabbrizzi, L.; Perotti, A.; Poggi, A.; Seghi, B.; Zanobini, F. (1987). "Dinickel and dicopper complexes with N,N-linked bis(cyclam) ligands. An ideal system for the investigation of electrostatic effects on the redox behavior of pairs of metal ions".Inorganic Chemistry 26 (21): 3527. doi:10.1021/ic00268a022. edit
- Jump up^ Davies, S. L.; Serradell, N.; Bolós, J.; Bayés, M. (2007). "Plerixafor Hydrochloride".Drugs of the Future 32 (2): 123. doi:10.1358/dof.2007.032.02.1071897. edit
- Jump up^ Davies, S. L.; Serradell, N.; Bolós, J.; Bayés, M. (2007). "Plerixafor Hydrochloride".Drugs of the Future 32 (2): 123. doi:10.1358/dof.2007.032.02.1071897. edit
- ^ Jump up to:a b &Na; (2007). "Plerixafor". Drugs in R & D 8 (2): 113–119. doi:10.2165/00126839-200708020-00006. PMID 17324009. edit
- ^ Jump up to:a b c d e Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. ISBN 3-85200-196-X.
- Jump up^ Wagstaff, A. J. (2009). "Plerixafor". Drugs 69 (3): 319. doi:10.2165/00003495-200969030-00007. PMID 19275275. edit
- ^ Jump up to:a b "CHMP Assessment Report for Mozobil". European Medicines Agency.
- Jump up^ Esté, J. A.; Cabrera, C.; De Clercq, E.; Struyf, S.; Van Damme, J.; Bridger, G.; Skerlj, R. T.; Abrams, M. J.; Henson, G.; Gutierrez, A.; Clotet, B.; Schols, D. (1999). "Activity of different bicyclam derivatives against human immunodeficiency virus depends on their interaction with the CXCR4 chemokine receptor". Molecular Pharmacology 55 (1): 67–73.PMID 9882699. edit
- Jump up^ Bridger, G.; et al. (1993). "Linked cyclic polyamines with activity against HIV. WO/1993/012096".
- Jump up^ Kalatskaya, I.; Berchiche, Y. A.; Gravel, S.; Limberg, B. J.; Rosenbaum, J. S.; Heveker, N. (2009). "AMD3100 is a CXCR7 Ligand with Allosteric Agonist Properties".Molecular Pharmacology 75: 1240. doi:10.1124/mol.108.053389.PMID 19255243. edit
- Jump up^ Cashen, A. F.; Nervi, B.; Dipersio, J. (2007). "AMD3100: CXCR4 antagonist and rapid stem cell-mobilizing agent". Future Oncology 3 (1): 19–27.doi:10.2217/14796694.3.1.19. PMID 17280498. edit
- Jump up^ "Mozobil approved for non-Hodgkin’s lymphoma and multiple myeloma" (Press release). Monthly Prescribing Reference. December 18, 2008. Retrieved January 3, 2009.
- Jump up^ Notice of Decision for MOZOBIL
- Jump up^ Smith, M. C. P.; Luker, K. E.; Garbow, J. R.; Prior, J. L.; Jackson, E.; Piwnica-Worms, D.; Luker, G. D. (2004). "CXCR4 Regulates Growth of Both Primary and Metastatic Breast Cancer". Cancer Research 64 (23): 8604–8612. doi:10.1158/0008-5472.CAN-04-1844. PMID 15574767. edit
- Jump up^ Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R. M.; Harsh, G. R.; Brown, J. M. (2010)."Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice". Journal of Clinical Investigation 120 (3): 694–705. doi:10.1172/JCI40283. PMC 2827954. PMID 20179352. edit
- Jump up^ Pitchford, S.; Furze, R.; Jones, C.; Wengner, A.; Rankin, S. (2009). "Differential Mobilization of Subsets of Progenitor Cells from the Bone Marrow". Cell Stem Cell 4 (1): 62–72. doi:10.1016/j.stem.2008.10.017. PMID 19128793. edit
- Jump up^ Wilson NM, Jung H, Ripsch MS, Miller RJ, White FA (March 2011). "CXCR4 Signaling Mediates Morphine-induced Tactile Hyperalgesia". Brain, Behavior, and Immunity 25(3): 565–73. doi:10.1016/j.bbi.2010.12.014. PMC 3039030. PMID 21193025.
- http://worlddrugtracker.blogspot.in/2013/11/plerixafor-new-treatment-approaches-for.html
External links
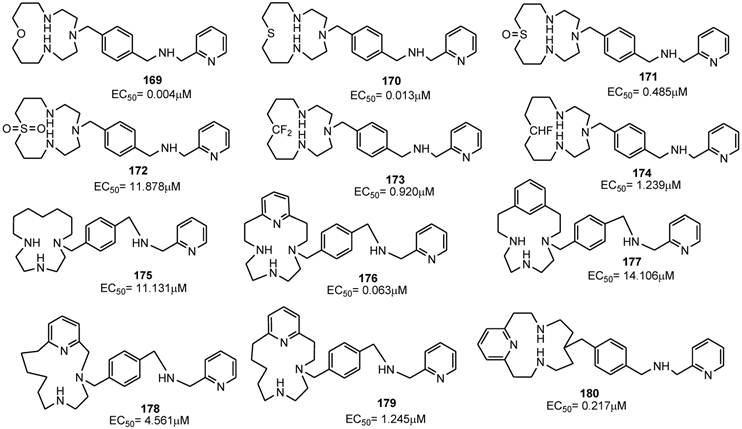
SEE ALSO..........http://www.scipharm.at/download.asp?id=1427
SEE..............https://www.academia.edu/5549712/2011531154034454SCHEME 15 IS SYNTHESIS OF PLERIXAFOR
SCHEME 15 IS SYNTHESIS OF PLERIXAFOR
read
ncur_powerpoint Courtney.ppt
faculty.swosu.edu/tim.hubin/share/ncur_powerpoint%20Courtney.ppt
... trials against cancer and for stem cell mobilization as “Mozobil” or “Plerixafor” ...NMR studies of AMD-3100 suggest that complex configuration is important.