
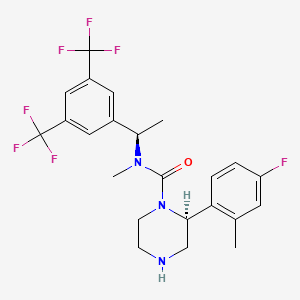
VESTIPITANT
(2S)-N-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl]-2-(4-fluoro-2-methylphenyl)-N-methylpiperazine-1-carboxamide
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-l- carboxylic acid [l-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl-amide
2-(S)-(4-fluoro-2-methylphenyl)piperazine-1-carboxylic acid [1-(R)-(3,5-bis-trifluoromethylphenyl)ethyl]methylamide (vestipitant)
Vestipitant [INN], UNII-S052TOI9BI, DCL001035,
CAS NO 334476-46-9
CAS NO 334476-46-9
Molecular Formula: C23H24F7N3O Molecular Weight: 491.444982
Elemental Analysis: C, 56.21; H, 4.92; F, 27.06; N, 8.55; O, 3.26
Vestipitant, also known as GW597599, is one of the most potent and selective NK(1) receptor antagonists ever discovered, showing appropriate pharmacokinetic properties and in vivo activity. Its actions support the utility of NK(1) receptor blockade in the alleviation of anxiety and, possibly, depression.
Vestipitant is a drug developed by GlaxoSmithKline which acts as a selective antagonist for the NK1 receptor. It is under development as a potential antiemetic and anxiolytic drug, and as a treatment for tinnitus.
Vestipitant mesylate is a tachykinin NK1 receptor antagonist in phase II clinical trials at GlaxoSmithKline for the treatment of postoperative nausea and vomiting. The drug candidate had been in clinical development at the company for several indications, including the treatment of tinnitus as monotherapy or in combination with paroxetine, the treatment of primary insomnia, the treatment of depression and anxiety and the treatment of chemotherapy-induced nausea and vomiting; however, no recent development has been reported for this research.
Vestipitant has anxiolytic properties and a good safety profile. Vestipitant was investigated for potential effect against chronic tinnitus as a stand-alone treatment and in conjunction with a selective serotonin reuptake inhibitor, paroxetine. No statistically significant treatment benefit effect was detected for tinnitus (intensity, pitch, and distress) VAS scores, arousal-anxiety VAS scores, Tinnitus Handicap Inventory, or tinnitus aggravation scores assessed on Days 1 and 14. However, a statistically significant worsening of tinnitus intensity and distress scores was observed after vestipitant compared with placebo for the mean data collected over the treatment period. No relevant differences in vestipitant plasma concentrations were observed between the subjects given the combination with paroxetine and those receiving vestipitant alone. No specific relationships were observed between tinnitus intensity and vestipitant plasma concentrations.
CONCLUSION: Although well-tolerated vestipitant, alone or in combination with paroxetine, was not effective in ameliorating tinnitus in this patient group.
Elemental Analysis: C, 56.21; H, 4.92; F, 27.06; N, 8.55; O, 3.26
Vestipitant, also known as GW597599, is one of the most potent and selective NK(1) receptor antagonists ever discovered, showing appropriate pharmacokinetic properties and in vivo activity. Its actions support the utility of NK(1) receptor blockade in the alleviation of anxiety and, possibly, depression.
Vestipitant is a drug developed by GlaxoSmithKline which acts as a selective antagonist for the NK1 receptor. It is under development as a potential antiemetic and anxiolytic drug, and as a treatment for tinnitus.
Vestipitant mesylate is a tachykinin NK1 receptor antagonist in phase II clinical trials at GlaxoSmithKline for the treatment of postoperative nausea and vomiting. The drug candidate had been in clinical development at the company for several indications, including the treatment of tinnitus as monotherapy or in combination with paroxetine, the treatment of primary insomnia, the treatment of depression and anxiety and the treatment of chemotherapy-induced nausea and vomiting; however, no recent development has been reported for this research.
Vestipitant has anxiolytic properties and a good safety profile. Vestipitant was investigated for potential effect against chronic tinnitus as a stand-alone treatment and in conjunction with a selective serotonin reuptake inhibitor, paroxetine. No statistically significant treatment benefit effect was detected for tinnitus (intensity, pitch, and distress) VAS scores, arousal-anxiety VAS scores, Tinnitus Handicap Inventory, or tinnitus aggravation scores assessed on Days 1 and 14. However, a statistically significant worsening of tinnitus intensity and distress scores was observed after vestipitant compared with placebo for the mean data collected over the treatment period. No relevant differences in vestipitant plasma concentrations were observed between the subjects given the combination with paroxetine and those receiving vestipitant alone. No specific relationships were observed between tinnitus intensity and vestipitant plasma concentrations.
CONCLUSION: Although well-tolerated vestipitant, alone or in combination with paroxetine, was not effective in ameliorating tinnitus in this patient group.
Vestipitant is a drug developed by GlaxoSmithKline which acts as a selective antagonist for the NK1 receptor. It is under development as a potentialantiemetic and anxiolytic drug,[1][2] and as a treatment for tinnitus.[3]
- Reddy, GK; Gralla, RJ; Hesketh, PJ (2006). "Novel neurokinin-1 antagonists as antiemetics for the treatment of chemotherapy-induced emesis". Supportive cancer therapy 3 (3): 140–2.doi:10.3816/SCT.2006.n.011. PMID 18632487.
- Brocco, M; Dekeyne, A; Mannoury La Cour, C; Touzard, M; Girardon, S; Veiga, S; De Nanteuil, G; Dejong, TR et al. (2008). "Cellular and behavioural profile of the novel, selective neurokinin1 receptor antagonist, vestipitant: a comparison to other agents". European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology 18 (10): 729–50.doi:10.1016/j.euroneuro.2008.06.002. PMID 18657401.
- ClinicalTrials.gov NCT00394056 Vestipitant Or Vestipitant/Paroxetine Combination In Subjects With Tinnitus And Hearing Loss
.........................
vestipitant
.........................
VESTIPITANT MESYLATE
CAS: 334476-64-1 of MESYLATE
- GW597588B
- UNII-OWR424W90Q
D06293, 334476-64-1
Journal of Thermal Analysis and Calorimetry, 2010 , vol. 102, 1 pg. 297 - 303
..........
INTRODUCTION

International patent application number WO2001/25219 describes piperazine derivatives. One such compound described therein is 2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-l- carboxylic acid [l-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl-amide (otherwise known as vestipitant) and it has the following chemical structure (I).

WO2001/25219 also describes the methanesulphonate salt of the compound (I).
The compound (I) and its pharmaceutically acceptable salts may be prepared by the processes described in International patent applications WO2001/25219 and WO2007/048642, which are incorporated herein by reference. Specifically, Examples 37 and 36 of WO2001/25219 describe the synthesis of the compound (I) as free base and as methanesulphonate salt respectively. Hydrochloride and acetate salts of the compound(I) are described in the Examples
38 and 18 respectively. Example 1 of WO2007/048642 discloses a process for preparing an intermediate in the synthesis of the compound(I).
...........................
Synthetic Process of Vestipitant
The following synthetic route was reported by Giuseppe Guercio et al from GlaxoSmithKline:
Org. Process Res. Dev., 2009, 13 (6), pp 1100–1110
DOI: 10.1021/op9002032
The initial chemical development synthetic route, derived from the one used by medicinal chemistry, involved several hazardous reagents, gave low yields and produced high levels of waste. Through a targeted process of research and development, application of novel techniques and extensive route scouting, a new synthetic route for GW597599 was developed. This paper reports the optimisation work of the third and last stage in the chemical synthesis of GW597599 and the development of a pilot-plant-suitable process for the manufacturing of optically pure arylpiperazine derivative 1. In particular, the process eliminated the use of triphosgene in the synthesis of an intermediate carbamoyl chloride, substantially enhancing safety, overall yield, and throughput.


1H NMR (600 MHz, DMSO-d6) δ 1.48 (d, J = 6.9 Hz, 3H); 2.31 (s, 3H); 2.39 (s, 3H); 2.74 (s, 3H); 2.95 (t, J = 12.2 Hz, 1H); 3.00−3.06 (m, 1H); 3.27 (dd, J = 12.5, 2.3 Hz, 1H); 3.28−3.35 (m, 1H); 3.38−3.43 (m, 1H); 3.47 (dt, J = 13.3, 3.1 Hz, 1H); 4.49 (dd, J = 11.8, 3.3 Hz, 1H); 5.35 (q, J = 6.5 Hz, 1H); 6.84 (td, J = 8.4, 2.6 Hz, 1H); 7.01 (dd, J = 10.2, 2.7 Hz, 1H); 7.29 (dd, J = 8.5, 6.0 Hz, 1H); 7.71 (bs, 2H); 8.02 (bs, 1H); 8.71 (bs, 1H); 9.02 (bs, 1H).
ES+: m/z 492 [MH − CH3SO3H]+, 341, 221; ES−: m/z 586 [M − H]−; 95 [CH3SO3]−.
13C NMR (150 MHz, DMSO-d6) δ 16.37, 18.81, 30.54, 39.79, 42.41, 45.70, 46.58, 52.41, 53.42, 112.48, 116.55, 121.02, 123.19 (d), 127.19, 127.44 (d), 130.34 (d), 134.00, 138.56, 144.79, 160.89, 163.2.
IR (Nujol mull, cm−1): 1653 (str. C═O), 1600 (str. C═C aromatic) (cm−1).
HPLC column type Betabasic C18; mobile phase A: buffer ammonium hydrogen carbonate 5 mM pH = 10/methanol 40/60% v/v and B buffer ammonium hydrogen carbonate 5 mM pH = 10/methanol 10/90% v/v; gradient: 0 min 100% A to 20 min 100% B. flow 1 mL/min; column temperature 40 °C; detector UV DAD @210 nm. Retention times 1: 13 min, purity >98%.
HPLC column type Chiralpack AD; mobile phase n-hexane/ethanol 86/14% v/v + 0.2% v/v purified water; flow 1 mL/min; column temperature 25 °C; detector UV DAD @210 nm. Retention time1: 4.56 min and opposite enantiomer 4.15 min, other diastereomers 5.20 and 14.2 min, respectively.
............................
SYNTHESIS
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-l- carboxylic acid [l-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl-amide
Preparation 1
(S)-2-(4-fluoro-2-methylphenyl)piperazine dihydrochloride
A suspension of (S)-3-(4-fluoro-2-methylphenyl)piperazin-2-one (S)-2-hydroxy-2- phenylacetate (14.0Kg; contains 16%w/w EtOAc hence 11.8 kg corrected for solvent) and tetrabutylammoniunn bromide (TBAB, 236g) in THF (94L) was warmed to 40°C to obtain a clear solution that was cooled to 30°C and then added to a slurry of sodium borohydride (powder grade, 5.5kg) in THF (41L) at 20°C, followed by THF (5.6L). The mixture was warmed to 35°C and then boron trifluoride-THF complex (36.6kg) was added over 90min, followed by THF (1L). The mixture was stirred for 6h and then IMS (47L) added over 3 hours. The mixture was distilled to ca. 94L, diluted with IMS (47L) and further distilled to 94L. The slurry was cooled to 25°C, filtered and the solids washed with IMS (2x35L). The combined filtrates were heated to 70°C and hydrogen chloride (5-6N in isopropanol, 15kg) added over 72min. The resulting slurry was heated at reflux for 3h, cooled to 20°C over 2h and then held at this temperature for 2h. The suspension was filtered, washed with IMS (3x24L) and the solids dried under vacuum at 45-50°C to give the title compound (6.87kg) as a white powder.
*H NMR NMR (D20) δ (ppm) 7.44 (dd, 1H), 7.03-7.00 (m, 2H), 4.89 (dd, 1H), 3.82-3.51 (m, 6H), 3.35 (s, 3H).
Preparation 2
(S)-tert-butyl3-(4-fluoro-2-methylphenyl)piperazine-l-carboxylate
hydrochloride
Triethylamine (5.5kg) was added to a slurry of (S)-2-(4-fluoro-2-methylphenyl)piperazine dihydrochloride (6.60kg, 94.6% assay) in EtOAc (38L) and was rinsed in with EtOAc (1L). The slurry was stirred at 40°C for 120 minutes and was then cooled to 20°C. 79.2%w/w Di-fe/ -butyl dicarbonate in EtOAc solution (6.29kg) was added over 60 minutes and was rinsed in with EtOAc (1L). The slurry was stirred for 15 minutes. Further 79.2%w/w di-fe/ -butyl dicarbonate in EtOAc solution (0.19kg) and EtOAc (1L) was added and the slurry was stirred for 43 minutes. EtOAc (5L), 79.2%w/w di-fe/ -butyl dicarbonate in EtOAc solution (0.25kg) and EtOAc (1L) were added and the slurry was then stirred for 15 minutes to complete the reaction. Water (18.7L) was added to dissolve all solids present and the lower aqueous layer was separated. The organic layer was washed with water (18.7L). The solution was distilled under reduced pressure to a total volume of 25L. Fresh EtOAc (37L) was added and the solution was distilled under reduced pressure to a total volume of 25L. EtOAc (49L) was added and the temperature was adjusted to 15°C. A slurry of the title compound (31.2g) in EtOAc (310ml) was added followed by 5.5M hydrogen chloride in isopropanol solution (0.412kg) rinsed in with EtOAc (1L). The mixture was stirred for 60 minutes to give a slurry. 5.5M Hydrogen chloride in isopropanol solution (3.6kg) was added portionwise over 55 minutes and was rinsed in with EtOAc (1L). The resultant slurry was stirred for 30 minutes at 15°C. The slurry was filtered and the solid was washed with EtOAc (2 x 16.8kg). The solid was dried under vacuum at 40°C to give the title compound (6.84kg) as a white solid.
*H NMR (500 MHz, DMSO-o^) δ ppm 9.89 (brs, 2 H), 7.88 (dd, 1 H), 7.13 - 7.20 (m, 2 H), 4.43 (d, 1 H), 4.07 (d, 1 H), 3.96 (d, 1 H), 3.30 - 3.38 (m, 2 H), 3.21 (m, 2 H), 2.39 (s, 3 H), 1.42 (s, 9 H). Preparation 3
(R)-l-(3,5-bis(trifluoromethyl)phenyl)-N-methylethanamine
To a suspension of (R)-l-(3,5-bis(trifluoromethyl)phenyl)-N-nnethylethanannine (S)-2- hydroxysuccinate (9Kg) in EtOAc (27L), 13% w/w aqueous sodium carbonate solution (27L) was added. The mixture was stirred for 30 minutes at 25°C to ensure complete dissolution. The layers were separated and the organic phase was washed with water (27L). EtOAc (36L) was added and the solution concentrated in vacuo to 18L. Further EtOAc (49Kg) was added and the solution concentrated in vacuo to 18L to give a colourless 33.4% w/w solution of the title compound in EtOAc (17.9Kg).
*H NMR for title compound (500 MHz, DMSO-i¼) δ ppm 8.01 (s, 2 H), 7.90 (s, 1 H), 3.79 (q, 7=6.56 Hz, 1 H), 2.35 (br s, 1 H), 2.10 (s, 3 H), 1.25 (d, 7=6.56 Hz, 3 H)
H NMR for EtOAc peaks (500 MHz, DMSO-i¼) δ ppm 4.02 (q, 7=7.17 Hz, 2 H), 1.98 (s, 3 H), 1.17 (t, 7=7.10 Hz, 3 H)
NMR shows a ratio of 1:6.1 the title compound: EtOAc.
Preparation 4
(S)-N-((R)-l-(3,5-bis(trifluoromethyl)phenyl)ethyl)-2-(4-fluoro-2- methylphenyl)-N-methylpiperazine-l-carboxamide methanesulfonate (Crystalline Form 1)
To a 33.4% w/w solution of (R)-l-(3,5-bis(trifluoromethyl)phenyl)-N-methylethanamine in EtOAc (14.70Kg) was added EtOAc (22L). The solution was vacuum purged three times with carbon dioxide gas and stirred under a flow of C02 at 20°C for 1 hour. Triethylamine (2.40Kg) was added followed by EtOAc (1.35Kg) and the solution stirred for 50 minutes under a flow of C02. Chlorotrimethylsilane (2.50Kg) was added over 30 minutes keeping the internal temperature below 25°C followed by EtOAc (1.35Kg) and the suspension stirred under a flow of C02 at 20°C for 30 minutes. Pyridine (2.85Kg) was added followed by EtOAc (2.70Kg). Thionyl chloride (3.25Kg) was added over 20 minutes followed by EtOAc (2.70Kg) and the suspension heated to 25°C for 6 hours. The reaction was cooled to 10°C and quenched with 28% w/w aqueous malic acid solution (14.30Kg). The layers were separated at 20°C and the organic phase washed with 14% w/w aqueous malic acid solution (13.50Kg), water (12.70Kg) and 20% w/w aqueous potassium phosphate dibasic solution (22.40Kg). EtOAc (4.50Kg) was added and the solution concentrated in vacuo to 15L. Further EtOAc (15L) was added and the solution concentrated in vacuo to 15L.
To the concentrated solution, EtOAc (5L) was added followed by (S)-tert-butyl 3-(4-fluoro-2- methylphenyl)piperazine-l-carboxylate hydrochloride (5.00Kg) and EtOAc (2.50Kg). Tributylamine (7.00Kg) was added and the suspension heated to reflux for 1 hour. The reaction was cooled to 30°C and EtOAc (27.20Kg) followed by water (15.00Kg) were added. The layers were separated, diethylamine (l.lOKg) was added to the organic phase and the solution heated to 40°C for 1 hour. The reaction was cooled to 30°C and washed with 0.5M sulfuric acid (25.90Kg), 0.5M sulfuric acid (15.45Kg) and water (15.00Kg).
To the organic phase, methanesulfonic acid (5.85Kg) was added and the solution heated to 40°C for 1 hour. The reaction was cooled to 10°C then 13%w/w aqueous ammonia solution (23.75Kg) was added over 30 minutes keeping the internal temperature below 35°C. The layers were separated at 30°C and the organic phase was washed with 1% w/w aqueous ammonia solution (15.15Kg) and water (15.00Kg). EtOAc (4.50Kg) was added to the organic phase and the solution was concentrated in vacuo to 15L. EtOAc (40L) was added and the solution concentrated in vacuo to 15L
Further EtOAc (10L) was added followed by methanesulfonic acid (1.20Kg) and (S)-N-((R)- l-(3,5-bis(trifluoromethyl)phenyl)ethyl)-2-(4-fluoro-2-methylphenyl)-N-methylpiperazine-l- carboxamide methansulfonate (25g) in isooctane (0.25Kg) and the suspension was stirred at 20°C for 70 minutes. Isooctane (50L) was added over 90 minutes and the reaction stirred for 1 hour. The suspension was filtered and washed with 2: 1 isooctane/EtOAc (12.5L) three times. The solid was co- milled to give the title compound (6.31Kg) as a white solid.
*H NMR (400 MHz, DMSO-i¾) δ ppm 8.96 (br. s., 2 H), 8.00 (s, 1 H), 7.71 (s, 2 H), 7.29 (dd,
7=8.56, 6.11 Hz, 1 H), 6.99 (dd, 7=10.27, 2.69 Hz, 1 H), 6.83 (td, 7=8.56, 2.45 Hz, 1 H), 5.35 (q, 7=6.60 Hz, 1 H), 4.52 (dd, 7=11.74, 3.18 Hz, 1 H), 3.52-3.22 (m, 4 H), 3.12-2.92 (m, 2 H), 2.74 (s, 3 H), 2.39 (s, 3 H), 2.37 (s, 3 H), 1.49 (d, 7=7.09 Hz, 3 H)
ES+: m/z 492 [MH - CH3S03H]+
Melt onset is 171°C obtained by Differential Scanning Calorimetry (DSC).
........................
SYNTHESIS
Example 36 2-(SM4-Fluoro-2-methyl-phenyl)-piperazine-1 -carboxylic acid M -(R)-
(3.5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide methansulphonate
To a suspension of intermediate 81 (4.9Kg) in AcOEt (137.2L), triethylamine (5.63L) was added. The mixture was cooled to 0°C then a solution of diterbuthyl dicarbonate (3.134Kg) in AcOEt (24.5L) was added in 35 min, maintaining the temperature between 0 and 5°C. The suspension was stirred at 0°C for 15 min, at 20/25°C for 1 hr, then washed with water (3 x 39.2L), concentrated to 24.5L and then added to a solution of triphosgene (1.97Kg) in AcOEt (24.5L) cooled to 0°C. Triethylamine (3.28L) was then added in 40 min, maintaining the temperature between 0 and 8°C. The suspension was stirred for 1 h and 45 min at 20/25°C and 30 min at 70Cand then the solution of intermediate 82 diluted with AcOEt (49L) and triethylamine (2.6L) was added in 30 min. The mixture was refluxed for 15 hrs.
The reaction mixture, cooled at 20/25°C was treated with aqueous solution of NaOH 10%v/v (36.75L). Organic phase was washed with HCI 4%v/v (46.55L) and NaCI 11 ,5%p/p (4 x 24.5L) then concentrated to 14.7L. and diluted with Ciclohexane (39.2L). The mixture was filtered through a silica pad (4.9Kg) that was washed twice with a mixture of CH/AcOEt 85/15 (2 x 49L). To the Eluted phases (14.7L) cooled at 20/25°C, methyl tertbutyl ether (49L) and methansulphonic acid (4.067L) were added. The mixture was washed with NaOH 10%v/v (31.85L) then with water (4 x 31.85L). Organic phase was concentrated to 9.8L, methyl tertbutyl ether (49L) was added and the solution filtered through a δmicron filter then concentrated to 9.8L. At 20/25°C MTBE (29.4L) and metansulphonic acid (1.098L) were added. The suspension was refluxed for 10 min, stirred at 20/25°C for 10hrs and 2 hrs at O°C.Then the precipitate was filtered, washed with methyl tertbutyl ether (4.9L) dried under vacuum at 20/25°C for 24 hrs to obtain the title compound (5.519Kg.) as white solid.
1H-NMR (DMSO) δ (ppm) 8.99 (bm, 1 H); 8.66 (bm, 1 H); 8.00 (bs, 1 H) 7.69 (bs, 2H); 7.27 (dd, 1 H); 7.00 (dd, 1 H); 6.83 (m, 1 H); 5.32 (q, 1 H) 4.47 (dd, 1 H); 3.50-3.20 (m, 4H); 2.96 (m, 2H); 2.72 (s, 3H); 2.37 (s, 3H) 2.28 (s, 3H); 1.46 (d, 3H). ES+: m/z 492 [MH - CH3SO3H]+ ES": m/z 586 [M - H]"; 95 [CH3SO3]"
Example 37
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-1 -carboxylic acid Ii -(R)- (3.5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide
To a solution of intermediate 40a (15.6g) in anhydrous THF (94ml), at 0°C, under N2, BH3THF 1 M/THF (154ml) was added. The solution was heated at reflux for 3 hr. HCI 37% (54ml) was slowly added maintaining the reaction mixture in an ice-bath and the reaction mixture was stirred at rt for 1 hr. Water was then added (125 ml) and solid NaHCO3 (62.4g) was added portionwise until a pH of 6.5.The aqueous phase was extracted with Et O (4x160 ml) and the combined organic extracts were dried over
Na2SO , the solids were filtered and evaporated to leave a colourless oil which was purified by flash chromatography (silica gel, EtOAc/Methanol 7/3). The obtained product was suspended in Et2O (220ml) and washed with NaHCO3 sat. (2x36ml). The combined organic phases were dried (Na2SO ) and evaporated to give the title compound as white foam (8.7g,). 1H-NMR (CDCI3) δ (ppm) 7.78 (s, 1 H); 7.60 (s, 2H); 7.28 (m, 1 H); 6.85 (dd, 1 H); 6.79 (td, 1 H); 5.53 (q, 1 H); 4.43 (dd, 1 H); 2.9-3.5 (m, 5H); 2.78 (m, 1 H), 2.71 (s, 3H); 2.43 (s, 3H); 1.47 (d, 3H).
Intermediate 40
2-(S)-(4-Fluoro-2-methyl-phenyl)-3-oxo-piperazine-1 -carboxylic acid ri-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide ( 40a ) 2-(S)-(4-Fluoro-2-methyl-phenyl)-3-oxo-piperazine-1 -carboxylic acid ri-(S)-(3.5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide.(-40b) To a solution of intermediate 39 (12.1g) in anhydrous DCM (270 mL), TEA (16.4 mL) was added. The solution was cooled down to 0°C and a solution of triphosgene (7.3 g) in anh. DCM (60 mL) was added drop-wise over 40 min. The reaction mixture was stirred at 0°C for 4 hr and was brought back to r.t. DIPEA (20.2 mL) was then added, followed by a solution of [1-(3,5- bis-trifluoromethyl-phenyl)-ethyl]-methyl-amine (23.6 g) in acetonitrile (300 mL) and an additional amount of acetonitrile (300 mL). The reaction mixture was warmed up to 95°C (oil bath T°C) without a water condenser to evaporate the DCM. When the internal temperature had reached 70°C, the flask was equipped with a water condenser, and the reaction mixture was heated at 70°C for an additional 2 hr (4 hr total). It was then brought back to r.t. and the solvent was evaporated. The residue was partitioned between DCM / 2% HCI and the phases were separated. The aqueous layer was extracted with DCM (1x) and the combined organic extracts were dried. The solids were filtered and the solvent evaporated to give a crude mixture of title compounds which were purified by flash chromatography (AcOEt/CH 8:2) to obtain the title compounds 40a (8.8 g) and 40 b (9.0 g) as white foams.
NMR (1H, DMSO-de): δ 8.16 (s, 1 H), 7.98 (s, 2H), 7.19 (dd, 1 H), 6.97 (dd, 1 H), 6.87 (td, 1 H), 5.34 (s, 1 H), 5.14 (q, 1 H), 3.45-3.2 (m, 4H), 2.53 (s, 3H), 2.27 (s, 3H), 1.56 (d, 3H).
Intermediate 40b: NMR (1H, DMSO-d6): δ 8.16 (s, 1 H), 7.95 (s, 2H), 7.19 (dd, 1 H), 6.98 (dd, 1 H), 6.90 (td, 1 H), 5.29 (q, 1 H), 5.28 (s, 1 H), 3.45-3.15 (m, 4H), 2.66 (s, 3H), 2.27 (s, 3H), 1.52 (d, 3H).
Intermediate 81 (S)-3-(4-Fluoro-2-methyl-phenyl)-piperazine dihydrochloride
To a solution of intermediate 39 (60.35g) in dry THF (180ml), at 0-3°C, under N2, BH3 THF 1 M/THF (1220mL) was added dropwise. The solution was refluxed for 4 hours then cooled to 0-3°C and methanol (240mL) was added. The reaction mixture was heated to room temperature then it was concentrated to dryness. The residue was redissolved in methanol (603.5mL), excess HCI 1 N in Et2O (1207mL) was added and the mixture was refluxed for 2 hours then cooled at 3°C for 4 hours. The suspension was filtered to obtain a white solid that was washed with Et2O (60.35mL) and dried to yield the title compound (72.02q)
1H-NMR (DMSO) δ (ppm) 11.0-9.5 (b, 4H); 7.99-7.19 (dd-m, 3H); 4.96 (dd, 1 H); 3.65-3.15 (m, 6H); 2.42 (s, 3H).
..................
HYDROCHLORIDE SALT
Example 38
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-1 -carboxylic acid |i -(R)-
(3,5-bis-trifluoromethyl-phenyl)-ethvπ-methyl-amide hydrochloride Example 37 (0.1 g) was dissolved in Ethyl Ether (0.8ml) at room temperature, then 1 M HCI solution in Ethyl Ether (0.6ml) was added. The suspension was stirred at 3°C for 3 hour, then filtered and washed with Ethyl Ether (1 ml) to afford the title compound ( 0.015g ) as a white solid. 1H-NMR (DMSO) δ (ppm) 9.31 (bm, 1 H); 9.11 (bm, 1 H); 8.02 (bs, 1 H); 7.72 (bs, 2H); 7.28 (dd, 1 H); 7.00 (dd, 1 H); 6.84 (m, 1 H); 5.34 (q, 1 H); 4.54 (dd, 1 H); 3.50-3.20 (m, 4H); 3.08 (m, 1 H); 2.93 (m, 1 H); 2.73 (s, 3H); 2.38 (s, 3H); 1.48 (d, 3H).
ACETATE SALT
Example 18
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-1 -carboxylic acid M -(R)-
(3.5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide acetate salt
To a solution of intermediate 40a (8.8 g) in dry THF (33 mL) under N2 BH3.THF (1 M solution in THF - 87 mL) was added and the reaction mixture was stirred at reflux for 3 hr, then cooled to r.t. and HCI (37%, 30 mL) was added drop-wise maintaining the reaction mixture in an ice-bath. The reaction mixture was stirred at r.t. for 1 hr. Water was then added (70 mL) and solid NaHCO3 (35.2 g) was added portion-wise until a pH of 6.5. The THF was evaporated and the aqueous phase was extracted with Et2θ (3 x 88 mL). The combined organic phases were dried, and evaporated to leave a colourless oil (7.37 g).
This crude oil was purified by flash chromatography (AcOEt/MeOH 7:3). The product obtained was suspended in Et2θ (125 mL) and washed with NaHCO3 sat. (2 x 20 mL). The clear combined organic phases were dried and evaporated to obtain the 2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine- 1 -carboxylic acid [1 -(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl- amide as white foam (5.27 g). This material (5.27 g) was dissolved in Et2θ (79 mL) and acetic acid (613 μL) was added drop-wise. The mixture was stirred at r.t. for 1 h and then at 0°C for 1h. The suspension was filtered to give the title compound (4.366 g) as a white solid. NMR (1H, DMSO-de): δ (ppm) 7.98 (s, 1 H), 7.70 (s, 2H), 7.87 (m, 1 H), 6.91 (m, 1 H), 6.77 (m, 1 H), 5.29 (q, 1 H), 4.23 (dd, 1 H), 3.2-2.6 (m, 6H), 2.68 (s, 3H), 2.3 (s, 3H), 1.89 (s, 3H), 1.48 (d, 3H). MS (m/z): 492 [M-CH3COO]+.
[ ]D = - 120.4°C Solvent (CHCI3); Source: Na; Cell volume [mL]: 1 ; Cell pathlength [dm]: 1 ; Cell temperature [°C]: 20; Wavelength [nm]: 589
'''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''
EXAMPLE 1 N-[1-(R) 3,5-bis-trifluoromethyl phenyl)-ethyl]-N-methyl carbamoyl chloride
[1-(R) 3,5-bis-trifluoromethyl phenyl)-ethyl]methyl amine L(−)maleate (13.5 g; 33.33 mmol) was suspended in ethyl acetate (39.9 ml) and ethanol (0.1 ml); aqueous sodium carbonate 13% (40 ml) was added and the mixture was stirred at a temperature 20-25° C. until a clear solution was formed. The water phase was discarded and the organic phase was washed with water (40 ml). Fresh ethyl acetate (49.87 ml) and ethanol (0.13 ml) were added, the solution was concentrated to 40 ml, a second amount of fresh ethyl acetate (49.87 ml) and ethanol (0.13 ml) was added and the solution was concentrated to 40 ml. Fresh ethyl acetate (109.7 ml) and ethanol (0.3 ml) were added under CO2 flow. A cycle of vacuum and CO2 in the vessel was applied, then CO2 was maintained for 10 minutes. Then, a neat Et3N (6.1 ml; 46.34 mmol) was added and the reaction mixture was stirred at a temperature 20-25° c. for 30 minutes. Trimethylmethylsilylchloride (6.4 ml; 40.42 mmol) was added in 30 minutes (exothermic step) and the reaction mixture was stirred for further 30 minutes at room temperature. Pyridine (5.4 ml; 66.66 mmol) was added, then SOCl2 (3.6 ml; 40.42 mmol) was added in 10 minutes. The reaction mixture was stirred at room temperature for 10 hours under CO2 atmosphere. 13% w/w aqueous racemic malic acid (60 ml) was added and the mixture was stirred for 15 minutes; the water phase was discarded then the organic phase was washed with water (60 ml); the water phase was discarded then the organic phase was washed with sodium carbonate 13% w/w (60 ml). Finally, the water phase was discarded and ethyl acetate (49.87 ml) and ethanol (0.13 ml) were added and the solution was concentrated to 50 ml; further ethyl acetate (49.87 ml) and ethanol (0.13 ml) were added and the solution was concentrated to dryness to give the title compound as a pale yellow (10.41 gr; 31.33 mmol 94% yield)
NMR-(d6-DMSO) δ (ppm)
8.04 δ (br s, 1H), 7.97 δ (br s, 2H), 5.52 δ (q, 1H), 2.97 δ (s, 3H), 1.66 δ (d, 3H)
EXAMPLE 2 (2R)-2-(4-fluoro-2-methylphenyl)-4-oxo-1-piperidinyl carbonyl; chloride
(2R)-2-(4-fluoro-2-methylphenyl)-4-oxo-1-piperidine L(−) mandelate (2 g; 5.57 mmol) was suspended in ethyl acetate (8 ml); aqueous sodium carbonate 13% w/w (10 ml) was added and the mixture was stirred at a temperature 20-25° C. until a clear solution was formed.
The water phase was discarded and the organic phase was washed with aqueous sodium chloride 10% w/w (4 ml). Fresh ethyl acetate (8 ml) were added, the solution was concentrated to 6 ml, a second amount of fresh ethyl acetate (8 ml) was added and the solution was concentrated to 6 ml.
Fresh ethyl acetate (2 ml) and neat Et3N (1.94 ml; 13.92 mmol) were added under CO2 flow at 0° C. The mixture was stirred for 10 minutes, then Trimethylmethylsilylchloride (1.42 ml; 11.14 mmol) was added in 5 minutes (exothermic step) and the reaction mixture was stirred for further 30 minutes at 0° C. Pyridine (0.58 ml; 7.24 mmol) was added, then SOCl2 (0.53 ml; 7.24 mmol) was added in 5 minutes. The reaction mixture was stirred at 0° C. for 1 h, then at a temperature 20-25° C. for 5 hours under CO2 atmosphere. Water (20 ml) was added was added; the water phase was discarded then the organic phase was washed with sodium carbonate 13% w/w (20 ml); the water phase was discarded then the organic phase was dried on sodium sulphate. The organic phase was filtered and concentrated to dryness to give the title compound as a pale yellow (1.5 gr; 5.57 mmol 100% yield)
HPLC Rt: 2.33 min; MS: [H+] 270
......................
J. Med. Chem., 2009, 52 (10), pp 3238–3247
DOI: 10.1021/jm900023b

a(a) (i) Mg, I2, THF, T = 70 °C, 2 h; (ii) LiBr, Cu2Br2, THF, room temp 1 h; (iii) CH3OCOCOCl, room temp, 2 h; (b) ethylenediamine, toluene, reflux, 6 h; (c) H2 (1 atm), 10% Pd/C, MeOH, 16 h; (d) (i) S-(+)-mandelic acid orR-(−)-mandelic acid, AcOEt, T = 3−5 °C, 2 h; (ii) filtration of the salt, then crystallization in AcOEt; (iii) 0.73 M NaOH; (e) (i) triphosgene, Et3N, CH2Cl2, T = 0 °C, 4 h; (ii) 1-[3,5-bis(trifluoromethyl)phenyl]ethyl]-N-methylamine, N(i-Pr)2Et, CH3CN, T = 70 °C, 2 h; (f) (i) 1 M BH3·THF, THF, reflux, 3 h; (ii) Et2O, AcOH.
.................
patents
WO 2012175434
WO 2008046882
WO 2004091624
WO 2004067093
...................
| WO1993005791A1 | Sep 18, 1992 | Apr 1, 1993 | Univ Pennsylvania | Prevention of hemolysis |
| WO2001025219A2 | Oct 5, 2000 | Apr 12, 2001 | Giuseppe Alvaro | Piperazine compounds |
| WO2004091624A1 * | Apr 16, 2004 | Oct 28, 2004 | Renzo Carletti | Combinations comprising paroxetine and 2- (s) - (4-fluoro-2-methyl-phenyl) -piperazine-1-carboxylic acid [1- (r)- (3,5-bis-trifluoro-2-methyl-phenyl) -ethyl]-methyl amide for treatment of depression and/or anxiety |
| WO2005082419A1 | Jan 6, 2005 | Sep 9, 2005 | Wayne Alan Boettner | Pharmaceutical compositions of neurokinin receptor antagonists and cyclodextrin and methods for improved injection site toleration |
| WO2007048642A1 | Oct 26, 2006 | May 3, 2007 | Ilaria Bientinesi | Process for preparing n, n-substituted carbamoyl halides |
| EP1897542A1 | Sep 7, 2006 | Mar 12, 2008 | Sanofi-Aventis | Aqueous formulation comprising an antitumor agent |
.......................................
1. Organic Process Research and Development, 2009 , vol. 13, 6 pg. 1100 - 1110
2. Magnetic Resonance in Chemistry, 2010 , vol. 48, 7 pg. 523 - 530
3. Org. Process Res. Dev., 2009, 13 (3), pp 489–493.
4. Synthesis of the NK1 receptor antagonist GW597599. Part 1: Development of a scalable route to a key chirally pure arylpiperazine
Org Process Res Dev 2008, 12(6): 1188.............
Org Process Res Dev 2008, 12(6): 1188.............
5.Journal of Thermal Analysis and Calorimetry, 2010 , vol. 102, 1 pg. 297 - 303
Di Fabio R, Alvaro G, Griffante C, Pizzi DA, Donati D, Mattioli M, Cimarosti Z, Guercio G, Marchioro C, Provera S, Zonzini L, Montanari D, Melotto S, Gerrard PA, Trist DG, Ratti E, Corsi M.
J Med Chem. 2011 Feb 24;54(4):1071-9. doi: 10.1021/jm1013264. Epub 2011 Jan 13.
Provera S, Guercio G, Turco L, Curcuruto O, Alvaro G, Rossi T, Marchioro C.
Magn Reson Chem. 2010 Jul;48(7):523-30. doi: 10.1002/mrc.2611.
Provera S, Martini L, Guercio G, Turco L, Costa L, Marchioro C.
J Pharm Biomed Anal. 2010 Nov 2;53(3):389-95. doi: 10.1016/j.jpba.2010.04.027. Epub 2010 Apr 29.
Sabbatini FM, Di Fabio R, Griffante C, Pentassuglia G, Zonzini L, Melotto S, Alvaro G, Capelli AM, Pippo L, Perdona' E, St Denis Y, Costa S, Corsi M.
Bioorg Med Chem Lett. 2010 Jan 15;20(2):623-7. doi: 10.1016/j.bmcl.2009.11.078. Epub 2009 Nov 20.
Di Fabio R, Griffante C, Alvaro G, Pentassuglia G, Pizzi DA, Donati D, Rossi T, Guercio G, Mattioli M, Cimarosti Z, Marchioro C, Provera S, Zonzini L, Montanari D, Melotto S, Gerrard PA, Trist DG, Ratti E, Corsi M.
J Med Chem. 2009 May 28;52(10):3238-47. doi: 10.1021/jm900023b.

 THANKS AND REGARD'S
THANKS AND REGARD'S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man's soul in action for you round the clock
need help, email or call me
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man's soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family