
Simeprevir
Inhibits HCV NS3/4A protease.
MEDIVIR ... originator
launched 2013
923604-59-5 CAS
C38H47N5O7S MF
749.93908 MW
IUPAC standard name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – ({7-methoxy-8-methyl-2-[4 - (propan-2-yl) -1,3-thiazol-2 -yl] quinolin-4-yl} oxy)-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
IUPAC traditional name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – {[2 - (4-isopropyl-1 ,3-thiazol-2-yl)-7-methoxy-8-methylquinolin-4- yl] oxy}-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – ({7-methoxy-8-methyl-2-[4 - (propan-2-yl) -1,3-thiazol-2 -yl] quinolin-4-yl} oxy)-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
IUPAC traditional name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – {[2 - (4-isopropyl-1 ,3-thiazol-2-yl)-7-methoxy-8-methylquinolin-4- yl] oxy}-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
- Olysio
- Simeprevir
- TMC 435
- TMC 435350
- TMC-435
- TMC435
- TMC435350
- UNII-9WS5RD66HZ
November 22, 2013 -- The U.S. Food and Drug Administration approved Olysio (simeprevir), a new therapy to treat chronic hepatitis C virus infection.
OLYSIO™ is the first once-daily protease inhibitor approved for the treatment of chronic hepatitis C in a combination antiviral regimen for adults with compensated liver disease
Hepatitis C is a viral disease that causes inflammation of the liver that can lead to diminished liver function or liver failure. Most people infected with the hepatitis C virus have no symptoms of the disease until liver damage becomes apparent, which may take several years. Most of these people then go on to develop chronic hepatitis C. Some will also develop scarring and poor liver function (cirrhosis) over many years, which can lead to complications such as bleeding, jaundice (yellowish eyes or skin), fluid accumulation in the abdomen, infections or liver cancer. According to the Centers for Disease Control and Prevention, about 3.2 million Americans are infected with the hepatitis C virus
Hepatitis C virus (HCV) infections affect approximately 3 percent of the worldwide population and often lead to cirrhosis and hepatocellular carcinoma. The standard therapy of pegylated- interferon and ribavirin induces serious side effects and provides viral eradication in less than 50% of patients. Combination therapy of HCV including ribavirin and interferonare currently is the approved therapy for HCV. Unfortunately, such combination therapy also produces side effects and is often poorly tolerated, resulting in major clinical challenges in a significant proportion of patients. Numerous direct acting agents (DAAs) have been or are being developed for treatment of HCV, such as telaprevir and boceprevir (both received MA approved in 2011 for use with interferon and ribavirin based therapy), however direct acting agents are linked to increased toxicity of treatment, the emergence of resistance, and to date do not provide a standard of care which is interferon free. The combination of direct acting agents can also result in drug-drug interactions. To date, no HCV therapy has been approved which is interferon free. There is therefore a need for new combination therapies which have reduced side effects, and interferon free, have a reduced emergence of resistance, reduced treatment periods and/or and enhanced cure rates.
Simeprevir (formerly TMC435) is an experimental drug candidate for the treatment of hepatitis C. It is being developed byMedivir and Johnson & Johnson‘s pharmaceutical division Janssen Pharmaceutica and is currently in Phase III clinical trials.[1]
Simeprevir is being tested in combination regimens with pegylated interferon alfa-2a and ribavirin,[3] and in interferon-free regimens with other direct-acting antiviral agents including daclatasvir[4] and sofosbuvir [5]
Simeprevir has been launched in 2013 in Japan by Janssen Pharmaceutical (JP) for use in combination with pegylated interferon (Peg-IFN) and ribavirin for the treatment of genotype 1 chronic hepatitis C virus (HCV) patients who are treatment naïve, prior non responders or relapsed following treatment with Peg-IFN with or without ribavirin. In 2013, the product has also been approved in the U.S. by Medivir and Janssen R&D Ireland for the oral treatment of chronic hepatitis C genotype 1 infection, in combination with peginterferon alfa and ribavirin in adults with compensated liver disease, including cirrhosis, who are treatment-naïve or who have failed previous interferon therapy (pegylated or non-pegylated) with ribavirin.
The drug candidate was originally developed at Medivir, which was acquired by Janssen R&D Ireland in 2012. In November 2004, Medivir entered into a license and research collaboration agreement with Tibotec, a Johnson & Johnson subsidiary, for the discovery and development of orally active protease inhibitors of the NS3/4A protease of HCV. In 2011, a codevelopment agreement between Pharmasset (now Gilead Sciences) and Tibotec was signed for the treatment of chronic hepatitis C (HCV) in combination with PSI-7977. Also in 2011, fast track designation was received in the U.S. for the treatment of chronic hepatitis C (CHC) genotype-1 infection.
In 2011, Tibotec Therapeutics, Division of Centocor Ortho Biotech Products, L.P. announced that it had changed its name to Janssen Therapeutics, Division of Janssen Products, LP.
“Hepatitis C is a complex disease and Janssen is committed to working with the HCV community, caregivers, and health care systems to address this global epidemic,” said Gaston Picchio, Hepatitis Disease Area Leader, Janssen Research & Development. “We are pleased that the FDA has granted simeprevir Priority Review, as it is a significant step forward in making this therapy available to physicians and their hepatitis C patients.”

Hepatitis C virus (HCV) is the leading cause of chronic liver disease worldwide.
Following initial acute infection, a majority of infected individuals develop chronic hepatitis because HCV replicates preferentially in hepatocytes but is not directly cytopathic. Chronic hepatitis can progress to liver fibrosis leading to cirrhosis, end- stage liver disease, and HCC (hepatocellular carcinoma), making it the leading cause of liver transplantations. This and the number of patients involved, has made HCV the focus of considerable medical research. Replication of the genome of HCV is mediated by a number of enzymes, amongst which is HCV NS3 serine protease and its associated cofactor, NS4A. NS3 serine protease is considered to be essential for viral replication and has become an attractive target for drug discovery.
Current anti-HCV therapy is based on (pegylated) interferon-alpha (IFN-α) in combination with ribavirin. Not only does this therapy result in a limited efficacy in that only part of the patients are treated successfully, but it also faces significant side effects and is poorly tolerated in many patients. Hence there is a need for further HCV inhibitors that overcome the disadvantages of current HCV therapy such as side effects, limited efficacy, poor tolerance, the emergence of resistance, as well as compliance failures.
Various agents have been described that inhibit HCV NS3 serine protease. WO05/073195 discloses linear and macrocyclic NS3 serine protease inhibitors with a central substituted proline moiety and WO 05/073216 with a central cyclopentyl moiety. Amongst these, the macrocyclic derivatives are attractive by overcoming one or more of the disadvantages of current anti-HCV therapy

(I) simeprevir
The compound of formula (I) is an inhibitor of the Hepatitis C virus (HCV) serine protease and is described in WO 2007/014926, published on 8 February 2007. This compound overcomes several of the disadvantages of current anti-HCV therapy and in particular shows pronounced activity against HCV, has an attractive pharmacokinetic profile, and is well-tolerated. Following the synthesis procedure described in Example 5 of WO 2007/014926, an amorphous solid form is obtained.
It now has been found that the compound of formula (I) can be converted into crystalline forms, which can advantageously be used as active ingredients in anti-HCV therapy. To that purpose, these crystalline forms are converted into pharmaceutical formulations.
.......................................................................................................
..............................
 simeprevir
simeprevir
OLYSIO (simeprevir) is an inhibitor of the HCV NS3/4A protease.
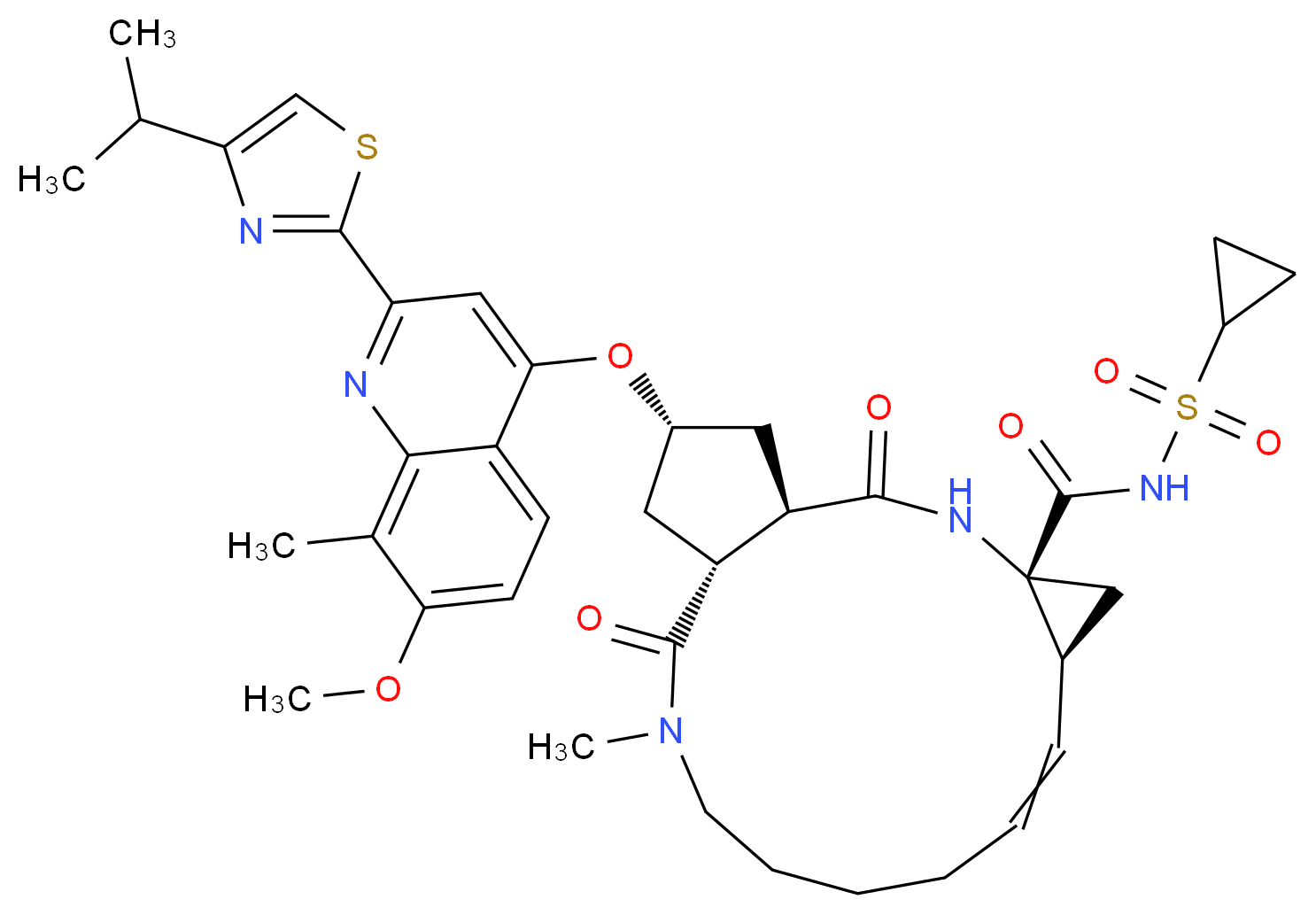
The chemical name for simeprevir is (2R,3aR,10Z,11aS,12aR,14aR)-N-(cyclopropylsulfonyl)-2[[2-(4-isopropyl-1,3-thiazol-2-yl)-7-methoxy-8-methyl-4-quinolinyl]oxy]-5-methyl-4,14-dioxo2,3,3a,4,5,6,7,8,9,11a,12,13,14,14atetradecahydrocyclopenta[c]cyclopropa[g][1,6]diazacyclotetradecine-12a(1H)-carboxamide. Its molecular formula is C38H47N5O7S2 and its molecular weight is 749.94. Simeprevir has the following structural formula:
 |
Simeprevir drug substance is a white to almost white powder. Simeprevir is practically insoluble in water over a wide pH range. It is practically insoluble in propylene glycol, very slightly soluble in ethanol, and slightly soluble inacetone. It is soluble in dichloromethane and freely soluble in some organic solvents (e.g., tetrahydrofuran and N,N-dimethylformamide).
OLYSIO (simeprevir) for oral administration is available as 150 mg strength hard gelatin capsules. Each capsule contains 154.4 mg of simeprevir sodium salt, which is equivalent to 150 mg of simeprevir. OLYSIO (simeprevir) capsules contain the following inactive ingredients: colloidal anhydrous silica, croscarmellose sodium, lactose monohydrate, magnesium stearate and sodium lauryl sulphate. The white capsule contains gelatin and titanium dioxide (E171) and is printed with ink containing iron oxide black (E172) and shellac (E904).
.................
Synthesis
Example 1 : preparation of 17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methyl- quinolin-4-yloxy]- 13-methyl-2, 14-dioxo-3, 13-diazatricyclo[ 13.3.0.04'6]octadec-7-ene- 4-carboxylic acid (16)
Synthesis of 4-hydroxy-2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinoline (6) Step 1 : synthesis of Λ/-(tert-butyloxycarbonyl)-3-methoxy-2-methylaniline (2)

1 2
Triethylamine (42.4 mL, 302 mmol) was added to a suspension of 3-methoxy-2- methylbenzoic acid (45.6 g, 274 mmol) in dry toluene (800 mL). A clear solution was obtained. Then, dppa (65.4 mL, 302 mmol) in toluene (100 mL) was slowly added. After 1 h at room temperature, the reaction mixture was successively heated at 500C for 0.5 h, at 700C for 0.5 h then at 1000C for 1 h. To this solution, t-BuOH (30.5 g, 411 mmol) in toluene (40 mL) was added at 1000C and the resulting mixture was refluxed for 7h. The solution was cooled to room temperature then successively washed with water, 0.5 N HCl, 0.5 N NaOH and brine, dried (Na2SO4), and evaporated to give 67 g of the target product: m/z = 237 (M)+.
_2: synthesis of 3-methoxy-2-methylaniline (3)

TFA (40.7 mL, 548 mmol) was added to a solution of jV-(teτt-butyloxycarbonyl)- 3-methoxy-2-methylaniline, in dichloro methane (500 mL). After 2 h at room temperature, TFA (40.7 mL, 548 mmol) was added and the resulting mixture was stirred at room temperature overnight. Then, volatiles were evaporated. The residue was triturated with toluene (100 mL) and diisopropylether (250 mL), filtered off and washed with diisopropyl ether (100 mL) to give 56.3 g of the title product as a TFA salt: m/z = 138 (M+H)+. The TFA salt was transformed to the free aniline by treatment with NaHCO3.
Step 3: synthesis of (2-amino-4-methoxy-3-methylphenyl)(methyl)ketone (4)

A solution Of BCl3 (1.0 M, 200 mL, 200 mmol) in CH2Cl2 was slowly added under nitrogen to a solution of 3-methoxy-2-methylaniline (26.0 g, 190 mmol) in xylene (400 mL). The temperature was monitored during the addition and was kept below 100C. The reaction mixture was stirred at 5°C for 0.5 h. Then, dry acetonitrile (13 mL, 246 mmol) was added at 5°C. After 0.5 h at 5°C, the solution was transferred into a dropping funnel and slowly added at 5°C to a suspension OfAlCl3 (26.7 g, 200 mmol) in CH2Cl2 (150 mL). After 45 min at 5°C, the reaction mixture was heated at 700C under a nitrogen stream. After evaporation Of CH2Cl2, the temperature of the reaction mixture reached 65°C. After 12 h at 65°C, the reaction mixture was cooled at 00C, poured onto ice (300 g), and slowly heated to reflux for 7h. After 2 days at room temperature, 6 N NaOH (50 mL) was added. The pH of the resulting solution was 2-3. The xylene layer was decanted. The organic layer was extracted with CH2Cl2. The xylene and CH2Cl2 layers were combined, successively washed with water, IN NaOH, and brine, dried (Na2SO4) and evaporated. The residue was triturated in diisopropyl ether at O0C, filtered off and washed with diisopropylether to give 13.6 g (40 %) of the title product as a yellowish solid: m/z = 180 (M+H)+.
Step 4: synthesis of 2'-[[(4-isopropylthiazole-2-yl)(oxo)methyl]amino]-4'-methoxy-3 '- methylacetophenone (5)

A solution of the compound 4 (18.6 g, 104 mmol) in dioxane (50 rnL) was added under nitrogen to a suspension of 4-isopropylthiazole-2-carbonyl chloride in dioxane (250 rnL). After 2 h at room temperature, the reaction mixture was concentrated to dryness. Then, the residue was partitioned between an aqueous solution of NaHCOs and AcOEt, organic layer was washed with brine, dried (Na2SO4), and evaporated. The residue was triturated in diisopropyl ether, filtered off and washed with diisopropyl ether to give 30.8 g (90 %) of the title product 5.
Step 5: synthesis of 4-hydroxy-2-(4-isopropylthiazole-2-yl)-7-methoxy-8- methylquinoline (6)

Potassium tert-butoxide (21.8 g, 195 mmol) was added to a suspension of the compound 5 (30.8 g, 92.7 mmol) in tert-butanol. The resulting reaction mixtures was heated at 1000C overnight. Then, the reaction mixture was cooled at room temperature and diluted with ether (100 mL). The precipitate was filtered off and washed with Et2O to give a powder (fraction A). The mother liquor was concentrated in vacuo, triturated in ether, filtered off, and washed with ether to give a powder (fraction 2). Fractions 1 and 2 were mixed and poured into water (250 mL). The pH of the resulting solution was adjusted to 6-7 (control with pH paper) with HCl IN. The precipitate was filtered off, washed with water and dried. Then, the solid was triturated in diisopropyl ether, fϊltered off and dried to give 26 g (88%) of the compound 6 as a brownish solid: m/z = 315 (M+H)+.
Synthesis of (hex-5-enyl)(methyl)amine (8)
O CF,
FX N Br' N O NH
H 7
(a) Sodium hydride (1.05 eq) was slowly added at 00C to a solution of JV-methyl- trifluoro-acetamide (25 g) in DMF (140 mL). The mixture was stirred for Ih at room temperature under nitrogen. Then, a solution of bromohexene (32,1 g) in DMF
(25 mL) was added dropwise and the mixture was heated to 700C for 12 hours. The reaction mixture was poured on water (200 mL) and extracted with ether (4 x 50 mL), dried (MgSO4), filtered and evaporated to give 35 g of the target product 7 as a yellowish oil which was used without further purification in the next step.
(b) A solution of KOH (187.7 g) in water (130 mL) was added dropwise to a solution of 7 (35 g) in methanol (200 mL). The mixture was stirred at room temperature for
12 hours. Then, the reaction mixture was poured on water (100 mL) and extracted with ether (4 x 50 mL), dried (MgSO4), filtered and the ether was distilled under atmospheric pressure. The resulting oil was purified by distillation under vacuum (13 mm Hg pressure, 500C) to give 7,4 g (34 %) of the title product 8 as a colourless oil: 1H-NMR (CDCl3): δ 5.8 (m, IH), 5 (ddd, J = Yl 2 Hz, 3.5 Hz, 1.8 Hz, IH), 4.95 (m, IH), 2.5 (t, J = 7.0 Hz, 2H), 2.43 (s, 3H), 2.08 (q, J= 7.0 Hz, 2H), 1.4 (m, 4H), 1.3 (br s, IH).
Preparation of 17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinolin-4-yloxyl- 13-methyl-2, 14-dioxo-3, 13-diazatricyclo[ 13.3.0.04'6loctadec-7-ene-4-carboxylic acid (16)

3-Oxo-2-oxa-bicyclo[2.2.1]heptane-5-carboxylic acid 9 (500 mg, 3.2 mmol) in 4 mL DMF was added at 00C to HATU (1.34 g, 3.52 mmol) and JV-methylhex-5-enylamine (435 mg, 3.84 mmol) in DMF (3 mL), followed by DIPEA. After stirring for 40 min at 00C, the mixture was stirred at room temperature for 5 h. Then, the solvent was evaporated, the residue dissolved in EtOAc (70 rnL) and washed with saturated NaHCOs (IO mL). The aqueous layer was extracted with EtOAc (2 x 25 mL). The organic phases were combined, washed with saturated NaCl (20 mL), dried (Na2SO4), and evaporated. Purification by flash chromatography (EtO Ac/petroleum ether, 2:1) afforded 550 mg (68%) of the target product 10 as a colorless oil: m/z = 252 (M+H)+.

A solution of LiOH (105 mg in 4 mlof water) was added at 00C to the lactone amide 10. After Ih, the conversion was completed (HPLC). The mixture was acidified to pH 2 - 3 with IN HCl, extracted with AcOEt, dried (MgSO4), evaporated, co-evaporated with toluene several times, and dried under high vacuum overnight to give 520 mg (88%) of the target product 11: m/z = 270 (M+H)+.

The l-(amino)-2-(vinyl)cyclopropanecarboxylic acid ethyl ester hydrochloride 12
(4.92 g, 31.7 mmol) and HATU (12.6 g, 33.2 mmol) were added to 11 (8.14 g,
30.2 mmol). The mixture was cooled in an ice bath under argon, and then DMF (100 mL) and DIPEA (12.5 mL, 11.5 mmol) were successively added. After 30 min at 00C, the solution was stirred at room temperature for an additional 3 h. Then, the reaction mixture was partitioned between EtOAc and water, washed successively with 0.5 N HCl (20 mL) and saturated NaCl (2 x 20 mL), and dried (Na2SO4). Purification by flash chromatography (AcOEt/CH2Cl2/Petroleum ether, 1 :1 :1) afforded 7.41 g (60%) of the target product 13 as a colorless oil: m/z = 407 (M+H)+.

DIAD (1.02 niL, 5.17 mmol) was added at -15°C under nitrogen atmosphere to a solution of 13 (1.5 g, 3.69 mmol), quinoline 6 (1.39 g, 4.43 mmol) and triphenyl- phosphine (1.26 g, 4.80 mmol) in dry THF (40 mL). After 4.5 h, at -15°C, the reaction mixture was partitioned between ice-cold water and AcOEt, dried (Na2SO4) and evaporated. The crude material was purified by flash column chromatography (gradient of petroleum AcOEt/CH2Cl2, 1 :9 to 2:8) to give 1.45 g (56 %) of the target product 14: m/z = 703 (M+H)+.

A solution of 14 (1.07 g, 1.524 mmol) and Hoveyda-Grubbs 1st generation catalyst (33 mg, 0.03 eq) in dried and degassed 1 ,2-dichloroethane (900 mL) was heated at 75°C under nitrogen for 12 h. Then, the solvent was evaporated and the residue purified by silica gel chromatography (25% EtOAc in CH2Cl2). 620 mg (60%) of pure macrocycle 15 were obtained, m/z = 674 (M+H)+. 1H NMR (CDCl3): 1.18-1.39 (m, 12H), 1.59 (m, IH), 1.70-2.08 (m, 5H), 2.28 (m, IH), 2.38 (m, IH), 2.62 (m, 2H), 2.68 (s, 3H), 2.83 (m, IH), 3.06 (s, 3H), 3.19 (sept, J= 6.7 Hz, IH), 3.36 (m, IH), 3.83 (m, IH), 3.97 (s, 3H), 4.09 (m, 2H), 4.65 (td, J= 4 Hz, 14 Hz, IH), 5.19 (dd, J= 4 Hz,
10 Hz, IH), 5.31 (m, IH), 5.65 (td, J= 4 Hz, 8 Hz, IH), 7.00 (s, IH), 7.18 (s, IH), 7.46
(d, J= 9 Hz, IH), 7.48 (s, IH), 8.03 (d, J= 9 Hz, IH).

A solution of lithium hydroxide (1.65 g, 38.53 mmol) in water (15 rnL) was added to a stirred solution of ester 15 (620 mg, 0.920 mmol) in THF (30 mL) and MeOH (20 mL). After 16 h at room temperature, the reaction mixture was quenched with NH4Cl sat., concentrated under reduced pressure, acidified to pH 3 with HCl IN and extracted with CH2Cl2, dried (MgSO4) and evaporated to give 560 mg (88%) of carboxylic acid 16. m/z = 647 (M+H)+. 1H NMR (CDCl3): 1.11-1.40 (m, 8H), 1.42-1.57 (m, 2H), 1.74 (m, 2H), 1.88-2.00 (m, 2H), 2.13 (m, IH), 2.28 (m, IH), 2.40 (m, IH), 2.59 (m, 2H), 2.67 (s, 3H), 2.81 (m, IH), 2.97 (s, 3H), 3.19 (m, IH), 3.31 (m, IH), 3.71 (m, IH), 3.96 (s, 3H), 4.56 (dt, J= 4 Hz, 12 Hz, IH), 5.23 (m, 2H), 5.66 (m, IH), 7.01 (s, IH), 7.10 (s, IH), 7.22 (d, J= IO Hz, IH), 7.45 (s, IH), 8.00 (d, J= 10 Hz, IH).
Example 2: Preparation of Λ/-[17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methyl- quinolin-4-yloxy]- 13-methyl-2, 14-dioxo-3, 13-diazatricyclo[ 13.3.0.04'6]octadec-7-ene- 4-carbonyll(cvclopropyl)sulfonamide (17) SIMEPREVIR

A solution of the compound 16 (560mg, 0.867 mmol) prepared according to Example 4, and carbonyldiimidazole (308 mg, 1.90 mmol) in dry THF (10 mL) was stirred at reflux under nitrogen for 2h. The reaction mixture was cooled to room temperature and cyclopropylsulfonamide (400 mg, 3.301 mmol) and DBU (286 mg, 1.881 mmol) were added. This solution was heated at 500C for 15 h. Then, the reaction mixture was cooled down at room temperature and concentrated under reduced pressure. The residue was partitioned between CH2Cl2 and HCl 1 N, the organic layer was washed with brine, dried (MgSO4) and evaporated. Purification by flash chromatography (gradient of EtOAc (0 to 25%) in CH2Cl2) afforded 314 mg of an off-white solid which was further washed with water, then isopropylether, and dried in the vacuum oven to deliver 282 mg (40%) of the pure title product 17, which is the compound of formula (I) SIMEPREVIR , as a white powder: m/z = 750 (M+H)+.
1H NMR (CDCl3): 0.99-1.52 (m, 14H), 1.64-2.05 (m, 4H), 2.77 (m, IH), 2.41 (m, 2H), 2.59 (m, 2H), 2.69 (s, 3H), 2.92 (m, 2H), 3.04 (s, 3H), 3.19 (m, IH), 3.40 (m, 2H), 3.98 (s, 3H), 4.60 (t, J= 13 Hz, IH), 5.04 (t, J= 11 Hz, IH), 5.37 (m, IH), 5.66 (m, IH), 6.21 (s, IH), 7.02 (s, IH), 7.22 (d, J= IO Hz, IH), 7.45 (s, IH), 7.99 (d, J= 10 Hz, IH), 10.82 (broad s, IH).
.....................
SYNTHESIS
Example 4: preparation of 17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methyl- quinolin-4-yloxy] - 13 -methyl-2, 14-dioxo-3 , 13 -diazatricyclo[ 13.3.0.04'6]octadec-7-ene- 4-carboxylic acid (46) FREE ACID
Synthesis of 4-hvdroxy-2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinoline (36) Step 1: synthesis of iV-(tert-butyloxycarbonyl)-3-methoxy-2-methylaniline (32)

31 32
Triethylamine (42.4 mL, 302 mmol) was added to a suspension of 3-methoxy-2- methylbenzoic acid (45.6 g, 274 mmol) in dry toluene (800 mL). A clear solution was obtained. Then, dppa (65.4 mL, 302 mmol) in toluene (100 mL) was slowly added. After 1 h at room temperature, the reaction mixture was successively heated at 50°C for 0.5 h, at 70°C for 0.5 h then at 100°C for 1 h. To this solution, t-BuOH (30.5 g, 411 mmol) in toluene (40 mL) was added at 100°C and the resulting mixture was refluxed for 7h. The solution was cooled to room temperature then successively washed with water, 0.5 N HCl, 0.5 N NaOH and brine, dried (Na2SO4), and evaporated to give 67 g of the target product: m/z = 237 (M)+.
Step 2: synthesis of 3-methoxy-2-methylaniline (33)

TFA (40.7 mL, 548 mmol) was added to a solution of iV-(tert-butyloxycarbonyl)-3- methoxy-2-methylaniline, in dichloromethane (500 mL). After 2 h at room temperature, TFA (40.7 mL, 548 mmol) was added and the resulting mixture was stirred at room temperature overnight. Then, volatiles were evaporated. The residue was triturated with toluene (100 mL) and diisopropylether (250 mL), filtered off and washed with diisopropyl ether (100 mL) to give 56.3 g of the title product as a TFA salt: m/z = 138 (M+H)+. The TFA salt was transformed to the free aniline by treatment with NaHCO3.
Step 3: synthesis of (2-amino-4-methoxy-3-methylphenyl)(methyl)ketone (34)

A solution OfBCl3 (1.0 M, 200 mL, 200 mmol) in CH2Cl2 was slowly added under nitrogen to a solution of 3-methoxy-2-methylaniline (26.0 g, 190 mmol) in xylene (400 mL). The temperature was monitored during the addition and was kept below 10°C. The reaction mixture was stirred at 5°C for 0.5 h. Then, dry acetonitrile (13 mL, 246 mmol) was added at 5°C. After 0.5 h at 5°C, the solution was transferred into a dropping funnel and slowly added at 5°C to a suspension OfAlCl3 (26.7 g, 200 mmol) in CH2Cl2 (150 mL). After 45 min at 5°C, the reaction mixture was heated at 70°C under a nitrogen stream. After evaporation Of CH2Cl2, the temperature of the reaction mixture reached 65°C. After 12 h at 65°C, the reaction mixture was cooled at 0°C, poured onto ice (300 g), and slowly heated to reflux for 7h. After 2 days at room temperature, 6 N NaOH (50 mL) was added. The pH of the resulting solution was 2-3. The xylene layer was decanted. The organic layer was extracted with CH2Cl2. The xylene and CH2Cl2 layers were combined, successively washed with water, IN NaOH, and brine, dried (Na2SO4) and evaporated. The residue was triturated in diisopropyl ether at O0C, filtered off and washed with diisopropylether to give 13.6 g (40 %) of the title product as a yellowish solid: m/z = 180 (M+H)+.
Step 4: synthesis of 2'-[[(4-isopropylthiazole-2-yl)(oxo)methyl]amino]-4'-methoxy-3 '- methylacetophenone (35)

A solution of (2-amino-4-methoxy-3-methylphenyl)(methyl)ketone (18.6 g, 104 mmol) in dioxane (50 mL) was added under nitrogen to a suspension of 4-isopropylthiazole-2- carbonyl chloride in dioxane (250 mL). After 2 h at room temperature, the reaction mixture was concentrated to dryness. Then, the residue was partitioned between an aqueous solution OfNaHCO3and AcOEt, organic layer was washed with brine, dried (Na2SO4), and evaporated. The residue was triturated in diisopropyl ether, filtered off and washed with diisopropyl ether to give 30.8 g (90 %) of the title product 35.
Step 5: synthesis of 4-hydroxy-2-(4-isopropylthiazole-2-yl)-7-methoxy-8- methylquinoline (36)

Potassium tert-butoxide (21.8 g, 195 mmol) was added to a suspension of 2'-[[(4-iso- propylthiazole-2-yl)(oxo)methyl]amino]-4'-methoxy-3'-methylacetophenone (35, 30.8 g, 92.7 mmol) in tert-butanol. The resulting reaction mixtures was heated at 100°C overnight. Then, the reaction mixture was cooled at room temperature and diluted with ether (100 mL). The precipitate was filtered off and washed with Et2O to give a powder (fraction A). The mother liquor was concentrated in vacuo, triturated in ether, filtered off, and washed with ether to give a powder (fraction 2). Fractions 1 and 2 were mixed and poured into water (250 mL). The pH of the resulting solution was adjusted to 6-7 (control with pH paper) with HCl IN. The precipitate was filtered off, washed with water and dried. Then, the solid was triturated in diisopropyl ether, filtered off and dried to give 26 g (88%) of the title product 36 as a brownish solid: m/z = 315 (M+H)+.
Synthesis of (hex-5-enyl)(methyl)amine (38)

Sodium hydride (1.05 eq) was slowly added at 0°C to a solution of iV-methyltrifluoro- acetamide (25 g) in DMF (140 mL). The mixture was stirred for Ih at room temperature under nitrogen. Then, a solution of bromohexene (32,1 g) in DMF (25 mL) was added dropwise and the mixture was heated to 70°C for 12 hours. The reaction mixture was poured on water (200 mL) and extracted with ether (4 x 50 mL), dried (MgSO4), filtered and evaporated to give 35 g of the target product 37 as a yellowish oil which was used without further purification in the next step.
Step B:
A solution of potassium hydroxide (187.7 g) in water (130 mL) was added dropwise to a solution of 37 (35 g) in methanol (200 mL). The mixture was stirred at room temperature for 12 hours. Then, the reaction mixture was poured on water (100 mL) and extracted with ether (4 x 50 mL), dried (MgSO4), filtered and the ether was distilled under atmospheric pressure. The resulting oil was purified by distillation under vacuum (13 mm Hg pressure, 50°C) to give 7,4 g (34 %) of the title product 38 as a colourless oil: 1H-NMR (CDCl3): δ 5.8 (m, IH), 5 (ddd, J= 17.2 Hz, 3.5 Hz, 1.8 Hz, IH), 4.95 (m, IH), 2.5 (t, J= 7.0 Hz, 2H), 2.43 (s, 3H), 2.08 (q, J= 7.0 Hz, 2H), 1.4 (m, 4H), 1.3 (br s, IH).
Preparation of 17-r2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinolin-4-yloxyl-
13-methyl-2,14-dioxo-3,13-diazatricvclori3.3.0.04'6loctadec-7-ene-4-carboxylic acid
£46}

3-Oxo-2-oxa-bicyclo[2.2.1]heptane-5-carboxylic acid 39 (500 mg, 3.2 mmol) in 4 mlDMF was added at 0°C to HATU (1.34 g, 3.52 mmol) and iV-methylhex-5- enylamine (435 mg, 3.84 mmol) in DMF (3 mL), followed by DIPEA. After stirring for 40 min at 0°C, the mixture was stirred at room temperature for 5 h. Then, the solvent was evaporated, the residue dissolved in EtOAc (70 mL) and washed with saturated NaHCO3 (10 mL). The aqueous layer was extracted with EtOAc (2 x 25 mL). The organic phases were combined, washed with saturated NaCl (20 mL), dried (Na2SO4), and evaporated. Purification by flash chromatography (EtOAc/petroleum ether, 2:1) afforded 550 mg (68%) of the target product 40 as a colorless oil: m/z = 252 (M+H)+.

A solution of LiOH (105 mg in 4 mlof water) was added at 0°C to the lactone amide 40. After Ih, the conversion was completed (HPLC). The mixture was acidified to pH 2 - 3 with IN HCl, extracted with AcOEt, dried (MgSO4), evaporated, co-evaporated with toluene several times, and dried under high vacuum overnight to give 520 mg (88%) of the target product 41: m/z = 270 (M+H)+.

The l-(amino)-2-(vinyl)cyclopropanecarboxylic acid ethyl ester hydrochloride 42 (4.92 g, 31.7 mmol) and HATU (12.6 g, 33.2 mmol) were added to 41 (8.14 g, 30.2 mmol). The mixture was cooled in an ice bath under argon, and then DMF (100 mL) and DIPEA (12.5 mL, 11.5 mmol) were successively added. After 30 min at 0°C, the solution was stirred at room temperature for an additional 3 h. Then, the reaction mixture was partitioned between EtOAc and water, washed successively with 0.5 N HCl (20 mL) and saturated NaCl (2 x 20 mL), and dried (Na2SO4). Purification by flash chromatography (AcOEt/CH2Cl2/Petroleum ether, 1:1:1) afforded 7.41 g (60%) of the target product 43 as a colorless oil: m/z = 407 (M+H)+.

DIAD (1.02 mL, 5.17 mmol) was added at -15°C under nitrogen atmosphere to a solution of 43 (1.5 g, 3.69 mmol), quinoline 36 (1.39 g, 4.43 mmol) and triphenyl- phosphine (1.26 g, 4.80 mmol) in dry THF (40 mL). After 4.5 h, at -15°C, the reaction mixture was partitioned between ice-cold water and AcOEt, dried (Na2SO4) and evaporated. The crude material was purified by flash column chromatography (gradient of petroleum AcOEt/CH2Cl2, 1 :9 to 2:8) to give 1.45 g (56 %) of the target product 44: m/z = 703 (M+H)+.

A solution of 44 (1.07 g, 1.524 mmol) and Hoveyda-Grubbs 1st generation catalyst (33 mg, 0.03 eq) in dried and degassed 1,2-dichloroethane (900 mL) was heated at 75°C under nitrogen for 12 h. Then, the solvent was evaporated and the residue purified by silica gel chromatography (25% EtOAc in CH2Cl2). 620 mg (60%) of pure macrocycle 45 were obtained, m/z = 674 (M+H)+. 1H NMR (CDCl3): 1.18-1.39 (m, 12H), 1.59 (m, IH), 1.70-2.08 (m, 5H), 2.28 (m, IH), 2.38 (m, IH), 2.62 (m, 2H), 2.68 (s, 3H), 2.83 (m, IH), 3.06 (s, 3H), 3.19 (sept, J= 6.7 Hz, IH), 3.36 (m, IH), 3.83 (m, IH), 3.97 (s, 3H), 4.09 (m, 2H), 4.65 (td, J= 4 Hz, 14 Hz, IH), 5.19 (dd, J= 4 Hz, 10 Hz, IH), 5.31 (m, IH), 5.65 (td, J= 4 Hz, 8 Hz, IH), 7.00 (s, IH), 7.18 (s, IH), 7.46 (d, J= 9 Hz, IH), 7.48 (s, IH), 8.03 (d, J= 9 Hz, IH).
Step F

A solution of lithium hydroxide (1.65 g, 38.53 mmol) in water (15 mL) was added to a stirred solution of ester 45 (620 mg, 0.920 mmol) in THF (30 mL) and MeOH (20 mL). After 16 h at room temperature, the reaction mixture was quenched with NH4Cl sat., concentrated under reduced pressure, acidified to pH 3 with HCl IN and extracted with CH2Cl2, dried (MgSO4) and evaporated to give 560 mg (88%) of carboxylic acid 46. m/z = 647 (M+H)+. 1H NMR (CDCl3): 1.11-1.40 (m, 8H), 1.42-1.57 (m, 2H), 1.74 (m, 2H), 1.88-2.00 (m, 2H), 2.13 (m, IH), 2.28 (m, IH), 2.40 (m, IH), 2.59 (m, 2H), 2.67 (s, 3H), 2.81 (m, IH), 2.97 (s, 3H), 3.19 (m, IH), 3.31 (m, IH), 3.71 (m, IH), 3.96 (s, 3H), 4.56 (dt, J= 4 Hz, 12 Hz, IH), 5.23 (m, 2H), 5.66 (m, IH), 7.01 (s, IH), 7.10 (s, IH), 7.22 (d, J= 10 Hz, IH), 7.45 (s, IH), 8.00 (d, J= 10 Hz, IH).
Example 5: Preparation of JV-ri7-r2-(4-isopropylthiazole-2-yl)-7-methoxy-8- methylquinolin-4- yloxyl - 13 -methyl-2, 14-dioxo-3 , 13 -diazatricyclol" 13.3.0.04'6loctadec- 7-ene-4-carbonyll (cvclopropyPsulfonamide (47) SIMEPREVIR

A solution of 17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinolin-4-yloxy]- 13-methyl-2, 14-dioxo-3, 13-diazatricyclo[l 3.3.0.04,6]octadec-7-ene-4-carboxylic acid 46 (560mg, 0.867 mmol) prepared according to Example 4, and carbonyldiimidazole (308 mg, 1.90 mmol) in dry THF (10 mL) was stirred at reflux under nitrogen for 2h. The reaction mixture was cooled to room temperature and cyclopropylsulfonamide (400 mg, 3.301 mmol) and DBU (286 mg, 1.881 mmol) were added. This solution was heated at 50°C for 15 h. Then, the reaction mixture was cooled down at room temperature and concentrated under reduced pressure. The residue was partitioned between CH2CI2 and HCl 1 N, the organic layer was washed with brine, dried (MgSO4) and evaporated. Purification by flash chromatography (gradient of EtOAc (0 to 25%) in CH2CI2) afforded 314 mg of an off-white solid which was further washed with water, then isopropylether, and dried in the vacuum oven to deliver 282 mg (40%) of the pure title product 47 SIMEPREVIR as a white powder: m/z = 750 (M+H)+.
1H NMR (CDCl3): 0.99-1.52 (m, 14H), 1.64-2.05 (m, 4H), 2.77 (m, IH), 2.41 (m, 2H), 2.59 (m, 2H), 2.69 (s, 3H), 2.92 (m, 2H), 3.04 (s, 3H), 3.19 (m, IH), 3.40 (m, 2H), 3.98 (s, 3H), 4.60 (t, J= 13 Hz, IH), 5.04 (t, J= 11 Hz, IH), 5.37 (m, IH), 5.66 (m, IH), 6.21 (s, IH), 7.02 (s, IH), 7.22 (d, J= 10 Hz, IH), 7.45 (s, IH), 7.99 (d, J= 10 Hz, IH), 10.82 (broad s, IH).
.......................
REFERENCES
- “Medivir Announces That Simeprevir (TMC435) Data Will Be Presented at the Upcoming AASLD Meeting”. Yahoo News. October 1, 2012. Retrieved November 6, 2012.
- Lin, TI; Lenz, O; Fanning, G; Verbinnen, T; Delouvroy, F; Scholliers, A; Vermeiren, K; Rosenquist, A et al. (2009). “In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor”. Antimicrobial agents and chemotherapy 53 (4): 1377–85. doi:10.1128/AAC.01058-08. PMC 2663092. PMID 19171797.
|displayauthors=suggested (help) - “Phase 3 Studies Show Simeprevir plus Interferon/Ribavirin Cures Most Patients in 24 Weeks”. hivandhepatitis.com. December 27, 2012.
- Medivir announces TMC435 in an expanded clinical collaboration. Medivir. 18 April 2012.
- Results from a phase IIa study evaluating Simeprevir and Sofosbuvir in prior null responder Hepatitis C patients have been presented at CROI. 6 March 2013.
- TMC-435350
Drugs Fut 2009, 34(7): 545 - Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350
Bioorg Med Chem Lett 2008, 18(17): 4853 - Synthesis of enantiomerically pure trans-3,4-substituted cyclopentanols by enzymatic resolution
Acta Chem Scand (1989) 1992, 46: 1127
PATENTS
- WO 2008092954
- WO 2007014926
- WO 2008092955
- WO 2000009543
- CN 102531932
- WO 2013061285
- WO 2011113859
- WO 2013041655
| WO2010097229A2 * | 26 Feb 2010 | 2 Sep 2010 | Ortho-Mcneil-Janssen Pharmaceuticals Inc | Amorphous salt of a macrocyclic inhibitor of hcv |
| WO2013037705A2 * | 7 Sep 2012 | 21 Mar 2013 | Fovea Pharmaceuticals | Aniline derivatives,their preparation and their therapeutic application |
| WO2005073195A2 * | 28 Jan 2005 | 11 Aug 2005 | Per-Ola Johansson | Hcv ns-3 serine protease inhibitors |
| WO2007014926A1 * | 28 Jul 2006 | 8 Feb 2007 | Tibotec Pharm Ltd | Macrocyclic inhibitors of hepatitis c virus |
The compound ritonavir, and pharmaceutically acceptable salts thereof, and methods for its preparation are described in WO94/14436. For preferred dosage forms of ritonavir, see US6,037, 157, and the documents cited therein: US5,484, 801, US08/402,690, and WO95/07696 and WO95/09614. Ritonavir has the following formula:
