Monday, 18 November 2013
Sunday, 17 November 2013
DOLASETRON, anti-emetic drug.

DOLASETRON
(3R)-10-oxo-8-azatricyclo[5.3.1.03,8]undec-5-yl 1H-indole-3-carboxylate, 115956-12-2 CAS

Dolasetron (trade name Anzemet) is a serotonin 5-HT3 receptor antagonist used to treat nausea and vomiting following chemotherapy. Its main effect is to reduce the activity of the vagus nerve, which is a nerve that activates the vomiting center in the medulla oblongata. It does not have muchantiemetic effect when symptoms are due to motion sickness. This drug does not have any effect on dopamine receptors or muscarinic receptors.
Dolasetron breaks down slowly, staying in the body for a long time. One dose usually lasts 4 to 9 hours and is usually administered once or twice daily. This drug is removed from the body by the liver and kidneys.
- Chemotherapy-induced nausea and vomiting
- 5-HT3 receptor antagonists are the primary drugs used to treat and prevent chemotherapy-induced nausea and vomiting. Many times they are given intravenously about 30 minutes before beginning therapy.
- Post-operative and post-radiation nausea and vomiting
- Is a possible therapy for nausea and vomiting due to acute or chronic medical illness or acute gastroenteritis
- Unlike most other 5HT3 antagonists, data is lacking for use of dolasetron with aprepitant in chemotherapy-induced nausea and vomiting (CINV).
- It is also sometimes used as an antiemetic (anti-vomiting medication) in veterinary medicine for dogs and cats.
Dolasetron is a well-tolerated drug with few side effects. Headache, dizziness, and constipation are the most commonly reported side effects associated with its use. There is a potential for prolonging of the QT interval to occur as well. There have been no significant drug interactions reported with this drug's use. It is broken down by the liver's cytochrome P450 system and it has little effect on the metabolism of other drugs broken down by this system. Intravenous dolasetron is contraindicated in Chemotherapy-induced nausea and vomiting (CINV). Doxorubicin and cyclophosphamide are as emetogenic as cisplatin, and preventive drugs should always be considered. The 5HT3 agonists are the mainstays of prevention and are frequently used in combination with other drugs such as corticosteroids and the NK1 receptor antagonist aprepitant. However, the FDA recently issued a drug communication stating that the injection form of dolasetron, a 5HT3 agonist, should no longer be used in adult or pediatric patients with CINV. Dolasetron injection can increase the risk of developing torsade de pointes, a potentially fatal abnormal heart rhythm. Patients with underlying heart conditions or existing heart rate or rhythm problems are at increased risk. Although the oral form of this agent can still be used, careful monitoring and correction of potassium and magnesium levels should be initiated prior to and during treatment. In addition, in older patients and in patients with heart failure, a slow heart rate, underlying cardiac disease, and those with renal impairment, monitoring with electrocardiography is indicated when this drug is used. Congenital long-QT syndrome and drugs that prolong the PR or QRS interval are contraindications to dolasetron therapy. Dolasetron injection may still be used for the prevention and treatment of postoperative nausea and vomiting. As per Food and Drug Administration.
- http://www.fda.gov/Drugs/DrugSafety/ucm237081.htm
- Katzung, Bertram G. Basic and Clinical Pharmacology, 9th ed. (2004). ISBN 0-07-141092-9
.............

ANZEMET (dolasetron mesylate) is an antinauseant and antiemetic agent. Chemically, dolasetron mesylate is (2α,6α,8α,9aβ)-octahydro-3-oxo-2,6-methano-2H-quinolizin-8-yl-lH- indole-3-carboxylate monomethanesulfonate, monohydrate. It is a highly specific and selective serotonin subtype 3 (5-HT3) receptor antagonist both in vitro and in vivo. Dolasetron mesylate has the following structural formula:
|
The empirical formula is C19H20N2O3 • CH3SO3H • H2O, with a molecular weight of 438.50.
Approximately 74% of dolasetron mesylate monohydrate is dolasetron base.
Dolasetron mesylate monohydrate is a white to off-white powder that is freely soluble in water and propylene glycol, slightly soluble in ethanol, and slightly soluble in normal saline.
ANZEMET Injection (dolasetron mesylate injection) is a clear, colorless, nonpyrogenic, sterile solution for intravenous administration. Each milliliter of ANZEMET Injection (dolasetron mesylate injection) contains 20 mg of dolasetron mesylate and 38.2 mg mannitol, USP, with an acetate buffer in water for injection. The pH of the resulting solution is 3.2 to 3.8.
ANZEMET Injection (dolasetron mesylate injection) multidose vials contain a clear, colorless, nonpyrogenic, sterile solution for intravenous administration. Each ANZEMET multidose vial contains 25 mL (500 mg) dolasetron mesylate. Each milliliter contains 20 mg dolasetron mesylate, 29 mg mannitol, USP, and 5 mg phenol, USP, with an acetate buffer in water for injection. The pH of the resulting solution is 3.2 to 3.7.
Synthesis of Dolasetron base is not very widely reported in literature. However, EP0266730/U.S. Pat. No. 4,906,755 describes process for the preparation endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate or Dolasetron mesylate, by the condensation of diethyl malonate with cis-1,4-dichloro-2-butene (2) in presence of lithium hydride in dimethylformamide to give diethyl-3-cyclopentene-1,1-dicarboxylate (3), which on hydrolysis and decarboxylation gave 3-cyclopentene-1-carboxylic acid (4). The compound (4) was further treated with thionyl chloride and pyridine in ethanol to obtain ethyl 3-cyclopentene-1-carboxylate (5). Compound (5) was oxidized to 4-ethoxycarbonyl-1,2-cyclopentanediol (6) by using N-methylmorpholine N-oxide in the presence of osmium tetroxide catalyst. The diol (6) was cleaved to the β-ethoxycarbonylglutaraldehyde (7) using sodium periodate and used directly in the next reaction. Robinson-Schopf cyclisation of the compound (7) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ethyl ester hydrochloride resulted in the pseudopelletierine derivative i.e. 7-ethoxycarbonyl-9-(ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-one (8). The ketone group of compound (8) was reduced with sodiumborohydride in ethanol to give 7-ethoxycarbonyl-9-(ethoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-ol (9). The reduced alcohol (9) was treated with dihydropyran to protect the hydroxyl group as a tetrahydropyranyl ether (10). Dieckmann cyclisation of the compound (10) using strong base (potassium t-butoxide) followed by aqueous acid hydrolysis and decarboxylation gave the desired alcohol. The resulting alcohols can exist in two conformations—axial and equatorial. The main product obtained by above procedure was the axial alcohol or endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (11) and it can be separated from the equatorial isomer by crystallization of the camphorsulfonate or tetrafluoroborate salt. The tetrafluoroborate salt of endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (11) was further reacted with 3-indolecarboxylic acid chloride in presence of silver tetrafluoroborate in anhydrous nitroethane at −78° C. to endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one or Dolasetron base, which was further converted into Dolasetron mesylate monohydrate (Scheme I) with a yield of 66%. No further purification is described.
The above process uses column chromatography for purification of compounds (9) and (10), which is expensive, time consuming and impractical on an industrial scale. The above patent does not disclose the yield and purity of Dolasetron mesylate obtained and so also for the intermediates. In addition, Osmium tetroxide used for preparation of compound (6) is toxic, has a corrosive action on eyes and hence difficult to use at industrial scale. Also this process uses high volume of water during preparation of the compound (8); preparation of compound (II) from compound (10) is tedious, because the workup involves several extractions with ethyl acetate and preparation of compound (I) in presence of silver tetrafluoroborate involves the use of expensive silver compound.

Another method described in EP0339669 provides a process for the preparation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one methanesulfonate or Dolasetron mesylate (1) by the condensation of dimethyl malonate with cis-1,4-dichloro-2-butene (2) in presence of lithium hydride in dimethyl formamide to give dimethyl-3-cyclopentene-1,1-dicarboxylate (12), which was decarbomethylated to obtain methyl-3-cyclopentene-1-carboxylate (13). This compound (13) was treated with m-chloroperbenzoic acid in dichloromethane to obtain 1-methoxycarbonyl-3-cyclopenteneoxide (14). The compound (13) on ozonolysis gave β-methoxycarbonylglutaraldehyde (15) or the epoxide (14) was reacted with periodic acid to obtain the β-methoxycarbonylglutaraldehyde (15), which was used directly in the next reaction. Robinson-Schopf cyclisation of the compound (15) with potassium hydrogen phthalate, acetonedicarboxylic acid and glycine ethyl ester hydrochloride gave the pseudopelletierine derivative i.e. 7-methoxycarbonyl-9-(methoxycarbonylethyl)-9-azabicyclo[3.3.1]nonan-3-one (16). The ketone group of compound (16) was reduced with sodiumborohydride in methanol to give 7-methoxycarbonyl-9-(methoxycarbonylmethyl)-9-azabicyclo-[3.3.1]nonan-3-ol (17). The reduced alcohol (17) was treated with dihydropyran to protect the hydroxyl group as a tetrahydropyranyl ether (18a) or treated with methylal to protect the hydroxyl group to obtain 3-methoxymethoxy-7-methoxycarbonyl-9-(methoxycarbonylmethyl)-9-azabicyclo[3.3.1]nonan-3-ol (18b).
Dieckmann cyclisation of the compound (18) using strong base (potassium t-butoxide) followed by aqueous acid hydrolysis and decarboxylation gave the endo-hexahydro-8-hydroxy-2,6-methano-2H-quinolizin-3-(4H)-one (11). The alcohol (11) was further reacted with 3-indolecarboxylic acid in presence of trifluoroacetic anhydride in dichloromethane to endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one or Dolasetron base (A), which was then converted into Dolasetron mesylate (1) (not shown in Scheme II) by treating with methanesulphonicacid in acetone (Scheme II).

Disadvantages of this process are:
- (i) use of high volume of water for preparation of compound (16) and
- (ii) preparation of compound (11) from compound (18) which is tedious because at the time of workup, ethyl acetate extractions take up longer period (20 h).
The process is not only time consuming but also expensive on an industrial scale. The patent does not disclose purity of Dolasetron base obtained nor for any of the intermediates.
The process as described in EP 0266730 involves treatment of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one (Dolasetron base) with a solution of methane sulfonic acid in ethanol to provide Dolasetron mesylate monohydrate. EP 0339669 describes crystallization of crude Dolasetron mesylate by dissolution in aqueous isopropanol and regeneration by adding ether. The polymorphic form obtained by the processes described in U.S. Pat. No. 4,906,755/EP 0266730 and EP 0339669 is designated herein as Dolasetron mesylate Form I.
The ability of the compound to exhibit more than one orientation or conformation of molecule within the crystal lattice is called polymorphism. Many organic compounds including active pharmaceutical ingredients (API's) exhibit polymorphism.
Drug substance existing in various polymorphic forms differs from each other in terms of stability, solubility, compressibility, flowability and spectroscopic properties, thus affecting dissolution, bioavailability and handling characteristics of the substance.
Rate of dissolution of an API's in patient's stomach fluid can have therapeutic consequences since it imposes an upper limit on the rate at which an orally administrated API can reach the patient bloodstream. Flowability affects the ease with which the material is handled while processing a pharmaceutical product.
Investigation of crystal polymorphism is an essential step in pharmaceutical research due to the influence of solid-state properties on dosage form.
As the polymorphs are known to possess different spectroscopic properties, technique such as X-Ray powder diffraction (XRPD), Fourier transformer Infrared (FT-IR) spectroscopy, Solid State 13C-NMR, and thermal method of analysis are keys to identify and characterize the new polymorphs or existing polymorphs.
The discovery of new polymorphs with same or better pharmaceutical equivalence and bioequivalence as that of the known polymorphs provides an opportunity to improve the performance characteristic of the pharmaceutical product.
The prior art describes isolation of endo-hexahydro-8-(3-indolylcarbonyloxy)-2,6-methano-2H-quinolizin-3(4H)-one or Dolasetron base as an oil. It is desirable to have the product in the solid form than oil, as solid is easy to handle and easy to purify.
Dolasetron base is isolated as a solid in EP 0339669. However, there is no evidence of polymorphism.
WO2006056081 discloses purification of Dolasetron base using strong acid especially methanesulphonic acid in presence of acid halide.

back to home for more updates


DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
Onrigin, Laromustine, VNP40101M, an alkylating agent under investigation for treating high-grade brain tumors.
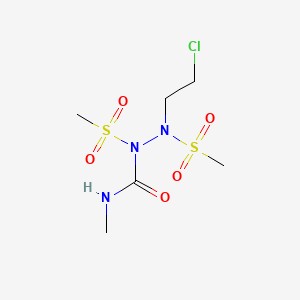 Cloretazine, Onrigin, VNP40101M, VNP-40101M, 101M, VNP 40101M, Laromustine [USAN], 173424-77-6 casno
Cloretazine, Onrigin, VNP40101M, VNP-40101M, 101M, VNP 40101M, Laromustine [USAN], 173424-77-6 casno
108736-35-22, (2-chloroethyl)-N-methyl-1,2-bis(methylsulfonyl)hydrazinecarboxamide
Molecular Formula: C6H14ClN3O5S2 Molecular Weight: 307.77546
 Vion Pharmaceuticals, Inc. was a New Haven, Connecticut based pharmaceutical company founded in March 1992 to commercialize several discoveries made in the biomedical laboratories at Yale University.
Vion Pharmaceuticals, Inc. was a New Haven, Connecticut based pharmaceutical company founded in March 1992 to commercialize several discoveries made in the biomedical laboratories at Yale University.
Vion Pharmaceuticals, Inc. was a New Haven, Connecticut based pharmaceutical company founded in March 1992 to commercialize several discoveries made in the biomedical laboratories at Yale University.
Two anticancer agents, Onrigin (laromustine), formerly cloretazine (VNP40101M), and Triapine, a ribunucloetide reductase inhibitor similar tohydroxyurea, were in human clinical trials. Onrigin (laromustine), a novel alkylating agent, was evaluated in a Phase 2 trial in elderly de novo poor-riskacute myeloid leukemia (AML). In addition, several trials of Onrigin (laromustine) were conducted in elderly patients with AML and myelodysplastic syndrome (MDS) in combination with cytarabine and in patients with brain tumors in combination with temozolomide. After Onrigin was rejected by the Food and Drug Administration for an AML indication in 2009 due to an unfavorable risk-benefit profile, the company went defunct.
Two anticancer agents, Onrigin (laromustine), formerly cloretazine (VNP40101M), and Triapine, a ribunucloetide reductase inhibitor similar tohydroxyurea, were in human clinical trials. Onrigin (laromustine), a novel alkylating agent, was evaluated in a Phase 2 trial in elderly de novo poor-riskacute myeloid leukemia (AML). In addition, several trials of Onrigin (laromustine) were conducted in elderly patients with AML and myelodysplastic syndrome (MDS) in combination with cytarabine and in patients with brain tumors in combination with temozolomide. After Onrigin was rejected by the Food and Drug Administration for an AML indication in 2009 due to an unfavorable risk-benefit profile, the company went defunct.

Onrigin


Subscribe to:
Posts (Atom)