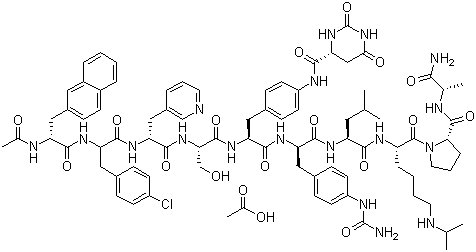
....................................
The synthesis of Degarelix has been described by solid phase synthesis using Boc chemistry (2001 :880 SYNTHLINE):
N-Boc-D-alanine (I) was coupled to the MBHA resin using diisopropyl carbodiimide and 1-hydroxybenzotriazole to afford resin (II). Subsequent cleavage of the Boc protecting group by means of trifluoroacetic acid (TFA) provided the D-alanine-bound resin (III). Sequential coupling and deprotection cycles were carried out with the following protected amino acids: N-Boc-L-proline (IV), N-alpha-Boc-N6-isopropyl-N6-carbobenzoxy-L- lysine (VI) and N-Boc-L-leucine (VIII) to afford the respective peptide resins (V), (VII) and (IX). N-alpha-Boc-D-4-(Fmoc-amino)phenylalanine (X) was coupled to (IX), yielding resin (XI). Cleavage of the side-chain Fmoc protecting group with piperidine DMF gave the aniline derivative (XII). This portion of the synthesis route is shown below in Scheme 1.
Scheme 1
After conversion to the corresponding urea by treatment with tert-butyl isocyanate, the Boc group was cleaved with TFA to produce resin (XIII). Further coupling with N-alpha- Boc-L-4-(Fmoc-amino)phenylalanine (XIV), followed by Fmoc deprotection with piperidine, furnished (XV). The aniline derivative (XV) was acylated with L-hydroorotic acid (XVI) to yield, after Boc group cleavage, resin (XVII). Coupling of (XVII) with N- Boc-L-serine(O-benzyl) (XVIII) and subsequent deprotection gave (XIX), as shown in Scheme 2, below:
Sche
Peptide (XIX) was sequentially coupled with N-alpha-Boc-D-(3-pyridyl)alanine (XX) and N-Boc-D-(4-chlorophenyl)alanine (XXII) to furnish, after the corresponding deprotection cycles with TFA, the resins (XXI) and (XXIII), respectively, as shown in Scheme 3, below:
Scheme 3
The coupling of resin (XXIII) with N-Boc-D-(2-naphthyl)alanine (XXIV) as before gave, after the corresponding deprotection cycle with TFA, resin (XXV). The peptide resin (XXV) was acetylated with Ac20 and finally deprotected and cleaved from the resin by treatment with HF to provide the target peptide, as shown in Scheme 4 below:
Scheme 4
Alternatively, after coupling of the peptide resin (XIII) with alpha-Boc-L-4-(Fmoc- amino)-phenylalanine (XIV), the Fmoc protecting group was not removed, yielding resin (XXVI). Subsequent coupling cycles with amino acids (XVIII), (XX), (XXII) and (XXIV) as above finally produced resin (XXVII). The Fmoc group was then deprotected by treatment with piperidine, and the resulting aniline was acylated with L-hydroorotic acid (XVI) to provide resin (XXVIII), as shown in Scheme 5 below:
Scheme 5
Resin (XXVIII) was finally cleaved and deprotected by treatment with HF, as shown in Scheme 6 below:
US 6214798 Bl, which describes a similar synthetic approach, suggests that classical peptide solution synthesis would be preferable for producing large quantities of product. The purity of the final material obtained in US 6214798 Bl is estimated, therein, to be about 98%, yet J Med. Chem. 2005, 48, 4851-4860 describing the same preparation yields a purity as measured by capillary zone electrophoresis (CZE) of 98%, which corresponds to 96% according to HPLC analysis.
Fmoc chemistry generally employs milder reaction conditions than the Boc chemistry, and thus can sometimes be less likely to cause the formation of by-products..
In addition, Fmoc chemistry does not employ hydrofluoric acid or other very strong acid (such as TFMSA or HBr/AcOH) is needed. Therefore these processes are much safer and environmentally friendly; factors that are important when engineering such a synthesis process on the large production scale.
In stepwise SPPS at least two types of protection for reactive functionalities other than those involved in peptide bond formation are necessary: temporary protection of the alpha-amino group, removable after formation of each peptide bond; and, side chain protection removable after assembly of the complete peptide chain.
When using Fmoc chemistry, a tBu protecting group is commonly used for the protection of the hydroxyl groups on Ser, Tyr and Thr residues. The tBu group is easily removed together with other side-chain protecting groups at the end of the peptide synthesis by using TFA.
WO2010121835 (*W0'835') describes the preparation of Degarelix using an Fmoc strategy and using tBu as the protecting group for the Ser residue. WO'835 also teaches that, unusually severe cleavage conditions such as long reaction time and 100% TFA as cleavage cocktail were required to successfully complete the final deprotection.. Such severe conditions are commonly known to result in increased side reactions, increased degradation of the peptide, and accordingly production of a lower quality of the resulting product.
One alternative to avoid using these severe deprotection conditions could be the use of different acid labile protecting group for Ser, such as Trt. However, it is well laiown in the art that the Trt group is very bulky and hydrophobic, and is frequently used to increase .the "steric hindrance" effect. See, e.g. Chem. Lett. 27, 1999 and J Pept. Sci. 2010; 16: 364-374, which describes that using the Trt protecting group in an Fmoc peptide synthesis could prevent formation of the target peptide due to "steric hindrance." The Degarelix sequence is inherently very hydrophobic. Therefore coupling of a large hydrophobic residue to the growing peptide chain is expected to cause slowing of the reaction and thus increased liability for the formation of impurities associated with deletions in the sequence and racemization. Synthesis efficiency is an important consideration when determining which general coupling reaction to use in an SPPS process. This efficacy can be limited by a number of different variables. One major variable that must be considered is the steric hindrance. This steric hindrance is determined by the nature of the peptide side chains and of their protecting groups. It is an important variable because it will allow the rapid acylation of the amino function and have a limiting effect on possible side reactions. See, e.g., ChemPep®, Fmoc Solid Phase Peptide Synthesis; Coupling reaction). Moreover, for the Degarelix synthesis, Fmoc-
Ser(Trt)-OH would be added to extremely bulky 4Aph(L-Hor) [4-[[[(4S)-hexahydro-2,6- dioxo-4-pyrimidinyl]carbonyl] amino] -L-phenylalanyl] residue. Accordingly, the use of this protecting residue would likely be quite difficult due to "steric hindrance". These "steric effects" are known to be of great importance when two molecules or two groups are in a close approach resulting in van der Waals forces repulsive effect. The repulsive potential can become quite large if the nonbonded distances are sufficiently short. This van der Walls repulsion can thus serve to slow a reaction, a situation that is typically refen-ed to as "steric hindrance" (Stereochemistry of Organic Compounds, E.L. Eliel et al, John Willey & Sons, 1994, 720 - 721 )
patents
WO 2011004260
WO 2011066386
WO 2012013331
WO 2012055903
WO 2012055905
WO 2013104745
WO 2013178788