MORRISVILLE, N.C.--(BUSINESS WIRE)--Oxygen Biotherapeutics, Inc., (NASDAQ: OXBT), a pharmaceutical company focused on developing drugs for the acute care market, today announced that it has selected Duke University’s Duke Clinical Research Institute, (DCRI) to conduct the Phase 3 trial of the Company’s newly acquired compound, levosimendan. DCRI is the world’s largest academic clinical research organization, with substantial experience in conducting cardiac surgery trials. The DCRI will serve as the coordinating center and Drs. John H. Alexander and Rajendra Mehta as lead investigators for the Phase 3 trial.
Levosimendan is a calcium sensitizer developed for intravenous use in hospitalized patients with acutely decompensated heart failure. The treatment is currently approved in more than 50 countries for this indication. The United States Food and Drug Administration (FDA) has granted Fast Track status for levosimendan for the reduction of morbidity and mortality in cardiac surgery patients at risk for developing Low Cardiac Output Syndrome (LCOS). In addition, the FDA has agreed to the Phase 3 protocol design under Special Protocol Assessment (SPA), and provided guidance that a single successful trial will be sufficient to support approval of levosimendan in this indication.
Oxygen Biotherapeutics recently acquired the North American rights to develop and commercialize levosimendan from Phyxius Pharma. It was discovered and developed by Orion Pharma, Orion Corporation of Espoo Finland.
John Kelley, CEO for Oxygen Biotherapeutics and former co-founder of Phyxius Pharma commented on this agreement: “We are extremely pleased to have an organization with the skill and expertise in conducting clinical trials that DCRI possesses as our partner. They have been responsible for managing a number of major cardiac surgery trials in the last decade, so their knowledge of this area of medicine is invaluable to us. At Phyxius Pharma, we worked with DCRI for the past three years to develop the clinical program for levosimendan. They played a key role in designing the Phase 3 protocol that has been approved by the FDA under SPA.”
According to the scientific literature, LCOS occurs in 5-10% of cardiac surgery patients, and is associated with a 10 – 15 fold increase in mortality. There is no drug currently approved for the prevention or treatment of LCOS. The Phase 3 clinical trial will study if levosimendan administered before and during surgery will reduce the incidence of LCOS and associated morbidity and mortality. There is substantial scientific evidence for the use of levosimendan in cardiovascular surgery, with over 25 published articles in peer reviewed journals and evidence of mortality reduction in some cardiac surgery trials of more than 50%.
The Phase 3 trial will be conducted in approximately 50 major cardiac surgery centers in North America. The trial will enroll patients undergoing coronary artery bypass graphs (CABG) and/or mitral valve surgery who are at risk for developing LCOS. The trial will be a double blind, randomized, placebo controlled study seeking to enroll 760 patients. It is expected that enrollment will begin in the third quarter of 2014, and will take approximately 18 months to complete. More details on the specifics of the trial will be released shortly.
John H. Alexander, M.D., MHS, Director of Cardiovascular Research, Duke Clinical Research Institute said about today’s announcement: “We are excited to be continuing our work on levosimendan and to move forward with the conduct of this innovative Phase 3 trial. The prevention of complications after high-risk cardiac surgery is clearly an area of unmet medical need. Our recent analysis of the work that has been published on levosimendan to date indicated a benefit to high-risk cardiac surgery patients at risk for developing LCOS. This next phase of research will be important to determine whether those results are verified in a large study. (Harrison et al. J Cardiothorac Vasc Anesth 2013;27:1224:32)”
About Oxygen Biotherapeutics
Oxygen Biotherapeutics, Inc. is developing medical products for the acute care market. The company recently acquired the North American rights to develop and commercialize levosimendan. The United States Food and Drug Administration (FDA) has granted Fast Track status for levosimendan for the reduction of morbidity and mortality in cardiac surgery patients at risk for developing Low Cardiac Output Syndrome (LCOS). In addition, the FDA has agreed to a Phase 3 protocol design under Special Protocol Assessment (SPA), and provided guidance that a single successful trial will be sufficient to support approval of levosimendan in this indication. The company also has developed a proprietary perfluorocarbon (PFC) therapeutic oxygen carrier called Oxycyte® that is currently in clinical and preclinical studies for intravenous delivery for indications such as traumatic brain injury, decompression sickness and stroke.
Levosimendan, which is the (−)-enantiomer of [[4-(1,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl)phenyl]hydrazono]propanedinitrile, and the method for its preparation is described in EP 565546 B1. Levosimendan is potent in the treatment of heart failure and has significant calcium dependent binding to troponin.
The hemodynamic effects of levosimendan in man are described in Sundberg, S. et al., Am. J. Cardiol., 1995; 75: 1061-1066 and in Lilleberg, J. et al., J. Cardiovasc. Pharmacol., 26(Suppl.1), S63-S69, 1995. Pharmacokinetics of levosimendan in man after i.v. and oral dosing is described in Sandell, E.-P. et al., J. Cardiovasc. Pharmacol., 26(Suppl.1), S57-S62, 1995. The use of levosimendan in the treatment of myocardial ischemia is described in WO 93/21921. Clinical studies have confirmed the beneficial effects of levosimendan in heart failure patients.The racemic mixture of [[4-(1,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl)phenyl]hydrazono]propanedinitrile (I) has been described earlier in the applicant's European Patent No. 38:3449 B1. It was shown that compound (I) is potent in the treatment of congestive heart failure and has significant calcium dependent binding to troponin.

Optically active enantiomers of (I) have been earlier described in the applicant's European Patent No. 565546 B1. It was shown that the cardiotonic potency is predominantly due to the (−) enantiomer of (I). A method for preparing pure (−) enantiomer of (I) using optically pure (−) enantiomer of 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone (II) as an intermediate compound was also disclosed.

The racemic compound (II) can be synthesized by methods known in the literature (J. Med. Chem., 17, 273-281 (1974)). The resolution of the racemic compound (II) has, however, been proved very difficult because the 4-amino group in the molecule is weakly basic. The salts of 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone with optically active acids hydrolyse on crystallization readily back to the compound (II) and to the resolving compound which interfere the resolution procedure or make it totally impossible.
The separation of the pure enantiomers of compound (II) on a chiral HPLC-column has been described in European patent application No. 208518. This method is, however, not applicable for industrial scale. An enantioselective seven step synthesis of (−)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone starting from (+)-2-chloropropionic acid has also been described in the literature (J. Org. Chem., 56, 1963 (1991)). The total yield in this method is only 12% giving (−)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone with an optical purity of 97.2%.
In the above mentioned European Patent No. 565546 B1 it was found that the racemic intermediate (II) can be resolved by treating (II) with L- or D-tartaric acid in excess in 2-propanol and recovering the diastereomeric crystalline salt. Optical purity of the product was further increased by dissolving the recovered basified product in dioxane. The racemic residue was crystallized from dioxane and the filtrate was evaporated to dryness yielding the desired pure enantiomer of the intermediate (II). The pure (−) enantiomer of (I) was prepared by treating (−) enantiomer of the intermediate (II) further with sodium nitrite and malononitrile in acidic conditions as described in the above mentioned European Patent No. 383449 B1.
Even if this process gives pure (−) enantiomer of (I), the necessity to use harmful dioxane limits its applicability in the large scale. Therefore there is a need for an improved process for preparing pure (−) enantiomer of (I).
Levosimendan is a highly potent cardiotonic that increases the sensitivity of the heart to calcium without causing a rise in intracellular calcium. The drug is marketed by Abbott under the trade name Simdax. It was first disclosed in U.S. Pat. No. 5,569,657.
The prior art indicates that pure levosimendan can be obtained by passing the racemic mixture over a chiral phase chromatography column. But the process becomes tedious and industrially unacceptable when a large quantity of material is involved.
One prior art technique involves using the optically-pure (−)-enantiomer of 6-(4-aminophenyl)-5-methylpyridazin-3-(2H)-one as starting material. The method of obtaining (−)-6-(4-aminophenyl)-5-methylpyridazin-3-(2H)-one is given in EP208518, which describes the separation of pure enantiomers of 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone using a chiral HPLC column.
CN1616437 describes treating (+)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone with 50% alkali or 50% acid.
JP10109977 discloses the use of 1-propanol/ethyl acetate as resolving solvent and L- or D-tartaric acid as resolving agent.
U.S. Pat. No. 5,569,657 discloses the preparation of levosimendan and its salts. (±)-6-(4-aminophenyl)-5-methylpyridazin-3-(2H)-one is dissolved in 2-propanol on heating. L-tartaric acid is gradually added to the solution and stirred on heating to obtain a clear solution. The solution is cooled slowly to room temperature and then stirred overnight at 200 C to obtain a crystalline product. On filtering, the wet salt is dissolved in water and to it potassium carbonate solution is added with stirring. The free base obtained is filtered, washed with water and dried. The product is further dissolved in dioxane on heating and allowed to cool to room temperature. The contents are filtered and dried under vacuum to obtain (−)-6-(4-aminophenyl)-5-methylpyridazin-3-(2H)-one crystalline solid. The pure (−)-6-(4-aminophenyl)-5-methylpyridazin-3-(2H)-one compound is then treated with sodium nitrite and malononitrile under acidic condition to obtain levosimendan. A disadvantage of the resolution process disclosed is that to obtain a high optical purity (99.5%) of the pyridazinone compound, recrystallisation with dioxane is required. Also the process involves multiple steps and is time consuming.
U.S. Pat. No. 6,180,789 describes the preparation of levosimendan by treating the (−)-enantiomer of 6-(4-amino phenyl)-5-methylpyridazin-3-(2H)-one, resolved using D- or L-tartaric acid in aqueous ethyl acetate, with sodium nitrite and malononitrile and further crystallizing with aqueous acetone. The patent also discloses other resolving agents such as benzoic acid, sulphuric acid, and resolving solvents such as isopropanol, isobutanol, isopropyl acetate, butyl acetate, acetone and acetonitrile. These conditions are said to cause partial resolution only.
There are certain drawbacks of the process disclosed in U.S. Pat. No. 6,180,789—
When D-tartaric acid is the resolving agent—
- Excess amount of resolving agent is required to achieve complete resolution.
- Seeding with D-tartaric acid salt of (−)-6-(4-amino phenyl)-5-methylpyridazin-3-(2H)-one is also needed in the process.
- Hot filtration of the precipitate is to be done; which is not at all workable when dealing with large batches at industrial scale.
- The temperature of reaction is to be maintained at 0° C.
- The enantiomeric purity of the product is low, even after giving a number of washings.
- In order to obtain the desired enantiomeric excess, it is necessary to perform recrystallisation with acetonitrile in the presence of absorbent followed by washing with excess acetonitrile. Treatment with a large amount of solvent increases the cost of process and also reduces the yield due to wastage.
When L-tartaric acid is the resolving agent—
- For the precipitation of salt, cooling up to −10° C. is required.
- The enantiomeric purity of the desired (−)-6-(4-amino phenyl)-5-methylpyridazin-3-(2H)-one product is quite low (78.7%).
The racemic mixture of [[4-(1,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl)phenyl]hydrazono]propanedinitrile (I) with melting point of 258 described earlier in applicant's patent application GB 2228004, which corresponds to U.S. Pat. No. 5,151,420. It was shown that the compound (I) is potent in the treatment of congestive heart failure and has significant calcium dependent binding to troponin. Our further studies have now unexpectedly revealed that the cardiotonic potency is predominantly due to the optically active (-) enantiomer of this compound. Furthermore it was found that the water solubility of the (-) enantiomer is over 30 fold compared to the racemate. The bioavailability of the (-) enantiomer was also found to be superior compared to racemate. Therefore the pure (-) enantiomer is especially suitable over the racemic compound to be used as a medicament for treating congestive heart failure.
...........................................
synthesis by rsolution of racemic starting matertial
synthesis by rsolution of racemic starting matertial

The optical resolution of 6 - (4-aminophenyl)-5-methyl-2 ,3,4,5-tetrahydropyridazin-3-one (I) with L-tartaric acid in isopropanol or D-tartaric acid in ethyl acetate gives the ( R)-enantiomer (II), which is then condensed with malonodinitrile (III) by means of NaNO2 and HCl in cool water.

Haikala, HO; Nore, PT; Honkanen, EJ; Pystynen, JJ; L Lian nberg, KK; Luiro, AM; Pippuri, AK (Orion Corporation); Heterocyclic cpds .. EP 0383449; JP 1990288868; US 5019575; US 5122524; US 5185332
GB 2251615; JP 1994504275; US 5424428; US 5512571; US 5569657; WO 9212135
WO 9735841
..........................................................................................
resolution using D Tartaric acid
100 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 2997 ml of ethyl acetate, 94,4 ml of water, 77,8 g of D-tartaric acid and 1.0 g of D-tartaric salt of (−)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone under nitrogen. The mixture was stirred in room temperature for 1.5 h. Thereafter the mixture was heated to 65° C. and stirred for 2 h. The precipitate was filtered hot and washed with 561 ml of ethyl acetate. The precipitate was mixed with 400 ml of water and pH of the mixture was adjusted to 9-10 with NH3. The mixture was cooled to 0° C. and stirred for 2 h. The precipitate was filtered, washed three times with 322 ml of cold water and dried in vacuum in 50° C. Yield was 35 g and the ratio of (−/+) enantiomers 93/7%. The product (35 g) was further added to 777 ml of acetonitrile and 2.0 g of celite under nitrogen. The precipitate was filtered hot and washed with 33 ml of acetonitrile which was added to the filtrate. 253 ml of acetonitrile was distilled from the filtrate and the remaining mixture was cooled to −5° C. The precipitate was filtered, washed with 76 ml of acetonitrile and dried in vacuum in 50° C. Yield 24.5 g. Ratio of (−/+) enantiomers 96/4%.
50 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 1500 ml of ethyl acetate, 46 ml of water, 37.5 g of D-tartaric acid and 1.0 g of D-tartaric salt of (−)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone. The mixture was stirred in room temperature for 1.5 h. Thereafter the mixture was heated to 65±3° C. and stirred for 3 h. The precipitate was filtered hot and washed with 116 ml of ethyl acetate of room temperature. The precipitate was mixed with 200 ml of water of room temperature and 44 g of potassium bicarbonate in 90 ml of water was slowly added. It was checked that pH was over 9.0. The mixture was cooled to 0±3° C. and stirred for 2 h. The precipitate was filtered, washed three times with 120 ml of cold water and dried in vacuum in 50±5° C. Yield 17.87 g. Ratio of (−/+) enantiomers 90.7/8.6%.
50 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 1500 ml of ethyl acetate, 45 ml of water and 37.3 g of L-tartaric acid. The mixture was heated to 60° C. and stirred for 2 h. The precipitate was filtered and the filtrate was cooled to −10° C. and kept in this temperature for 2 h. The precipitate that crystallized from the filtrate was filtered and dried in vacuum in 50° C. The precipitate was mixed with 200 ml of water in room temperature and 43 g of potassium bicarbonate in 90 ml of water was slowly added. It was checked that pH was over 9.0. The mixture was cooled to 0° C. and stirred for 2 h. The precipitate was filtered, washed three times with 120 ml of cold water and dried in vacuum in 50±5° C. Yield 20.61 g. Ratio of (−/+) enantiomers 78.7/21.2%.
1 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 30 ml of isopropanol and 0.6 g of benzoic acid. The mixture was boiled until dissolved and cooled to room temperature whereupon the product crystallized. The crystalline product was filtered and the ratio of the benzoic acid salts of the enantiomers was determined. Ratio of (−/+) enantiomers 74.1/25.5%.
1 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 30 ml of isopropanol and 0.48 g of concentrated sulphuric acid. The mixture was boiled and cooled to room temperature. The crystalline product was filtered and the ratio of the sulphate salts of the enantiomers was determined. Ratio of (−/+) enantiomers 65.1/34.9%
5 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 75 ml of ethyl acetate, 3.1 ml of water and 3.73 g of L-tartaric acid. The mixture was boiled for 3.5 h, the precipitate was filtered and the filtrate was cooled to −10° C. The precipitate that crystallized from the filtrate was filtered and dried in vacuum in 50° C. Yield 2.86 g. The ratio of the L-tartrate salts of the enantiomers (−/+) 72/27%.
1 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 20 ml of isobutanol and 0.75 g of L-tartaric acid. The mixture was boiled and cooled. A sample was taken as soon as the crystallization started (at 64° C). The ratio of the L-tartrate salts of the enantiomers (−/+) 53/46%. 0.6 ml of water was added to the mixture, the mixture was boiled, cooled and a sample was taken at the beginning of the crystallization (at 64° C.). The ratio of the L-tartrate salts of the enantiomers (−/+) 60/40%. Again 0.6 ml of water was added to the mixture and the previous procedure was repeated. The product started crystallize at 46° C. The ratio of the L-tartrate salts of the enantiomers (−/+) 56/44%.
1 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 60 ml of isopropyl acetate and 0.75 g of L-tartaric acid. The mixture was boiled and a sample was taken from the undissolved precipitate. The ratio of the L-tartrate salts of the enantiomers (−/+) 44/56%. 1.2 ml of water was added to the mixture, whereupon the precipitate dissolved. The mixture was cooled and a sample was taken at the beginning of the crystallization (at 68° C.). The ratio of the L-tartrate salts of the enantiomers (−/+) 24/76%.
1 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 50 ml of ethyl acetate, 0.75 g of L-tartaric acid and A) 0.5, B) 1.0 or C) 1.5 ml of water. The mixture was boiled and cooled. The mixture was filtered and a sample was taken from the precipitate and from the filtrate at the beginning of the crystallization. The ratio of the L-tartrate salts of the enantiomers (−/+) %:
| Precipitate | Filtrate | Crystallization temperature, ° C. | |||
| A) | 29/71 | 66/30 | 52 | ||
| B) | 22/77 | 65/32 | 54 | ||
| C) | 20/80 | 85/13 | 50 | ||
1 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 50 ml of butyl acetate and 0.75 g of L-tartaric acid. The mixture was boiled and cooled. The mixture was filtered and a sample was taken from the precipitate at the beginning of the crystallization (64° C.). The ratio of the L-tartrate salts of the enetntiomers (−/+) 44/55%.
1 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 10 ml of acetone and 0.75 g of L-tartaric acid. The mixture was warmed until dissolved (54° C.) and cooled to 0° C. A sample was taken from the precipitate. The ratio of the L-tartrate salts of the enantiomers (−/+) 49/42%.
1 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone was added to 44 ml of acetonitrile and 0.75 g of L-tartaric acid. The mixture was boiled and cooled. A sample was taken from the precipitate at the beginning of the crystallization. The ratio of the L-tartrate salts of the enantiomers (−/+) 43/50%.
The 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone obtained in Example 2 with (−/+) resolution % of 96/4 was treated with sodium nitrite and malononitrile as described in the European Patent No. 383449 B1. 10 g of the recovered [[4-(1,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl)phenyl]hydrazono]propanedinitrile with (−/+) resolution % of 96/4 was added to 150 ml of acetone, 0.9 ml of water, 0.2 g of activated carbon and 0.4 g of Celite. The mixture was refluxed for 1 h and filtered hot. The precipitate was washed with 10 ml of hot acetone which was added the to the filtrate. The filtrate was refluxed for 30 min. 61 ml of acetone was distilled from the filtrate and the remaining mixture was cooled to 0-(−5) ° C. The mixture was filtered and washed with 10 ml of cold acetone. The crystalline product was dried in vacuum in 50° C. The product contained over 99% of the desired (−) enantiomer and the yield was 6.8 mg. The product was substantially pure crystalline polymorphic form I.
..........................................................
use of di-p-anisoyl-D-tartaric acid as resolution agents
use of di-p-anisoyl-D-tartaric acid as resolution agents

di-p-anisoyl-D-tartaric acid of formula III is used.


(a) resolving racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone of Formula II,

with di-p-anisoyl-D-tartaric acid of formula III in the presence of a mixture of water and ethanol to form the diastereomeric (−)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone di-p-anisoyl-D-tartaric acid salt of formula VI,

typically at a temperature ranging from 55° C. to 70° C.;
(b) treating the diastereomeric salt of formula VI with an organic or inorganic base selected from ammonia, sodium methoxide, sodium hydroxide, potassium hydroxide, sodium carbonate, potassium bicarbonate, potassium carbonate or sodium bicarbonate to obtain the (−)-isomer of 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone of formula V;

(c) reacting (−)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone of formula V with sodium nitrite and malononitrile under acidic conditions to obtain levosimendan of formula I.

50 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone and 500 ml ethanol were added to a flask and stirred at 65° C. for 30 minutes. Further di-p-anisoyl-D-tartaric acid (113.3 g) and 500 ml water were added and stirring was continued for 1 hour. The reaction mass was cooled to 15° C. and stirred for another 30 minutes. Then the mass was filtered, washed with water and dried under vacuum at 55° C. to obtain the corresponding diastereomeric salt (yield—110 g, purity—99.5%).
The obtained salt and water (500 ml) were added to a reaction vessel and stirred at 25-30° C. for 30 minutes. The pH of the resulting solution was adjusted to 8-9 by adding ammonia solution and stirring was continued for 30 minutes. After completion of reaction, the contents were filtered and dried under vacuum at 55° C. to obtain solid (−)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone (yield—24 g, purity—99.6%).
To a solution of (−)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone (20 g) and dilute hydrochloric acid (52 ml of concentrated hydrochloric acid in 789 ml of water), 8 g of dilute sodium nitrite (8 g of sodium nitrite in 52 ml of water) was added and stirred. After 10 minutes, malononitrile solution (6.3 g malononitrile in 52 ml of water) was added. The solution was stirred for 1 hour at room temperature. The pH of the suspension was adjusted to 6.0 with sodium acetate solution. The suspension was filtered, washed with water followed by ethanol and then dried to obtain solid levosimendan (yield—22 g, purity—99.9%).
10 g of racemic 6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone and 100 ml ethanol were added and stirred at 65° C. for 30 minutes. Di-p-anisoyl-L-tartaric acid (22.6 g) and 100 ml water were added and stirring continued for 1 hour. After completion of reaction, the mass was cooled to 15° C. to obtain a solid and then filtered. The mother liquor obtained was concentrated to remove ethanol then basified with ammonia. The precipitated solid was filtered and treated with sodium nitrite and malononitrile as described above to obtain solid levosimendan (yield—4 g, purity—99.6%).
25 g of (+)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3-(2H)-pyridazinone, obtained from step 1 of Example 1, and 50% sodium hydroxide solution were heated at 90° C. The reaction mass was cooled to room temperature, filtered and washed with water. The solid obtained was charged in a flask along with 250 ml ethanol and stirred at 65° C. for 30 minutes. Di-p-anisoyl-D-tartaric acid (56.6 g) and 250 ml water were added and stirring was continued for 1 hour. The reaction mass was cooled to 15° C. and stirred for another 30 minutes. The reaction mass was filtered and washed with water. The wet solid diastereomeric salt was charged in a round bottom flask along with water and the contents stirred at 25° C. To this mixture, 10% potassium carbonate was added and the pH was adjusted to 8-9. The reaction mass was stirred for 30 minutes and then filtered. The solid obtained was treated with sodium nitrite and malononitrile solution in presence of dilute hydrochloric acid to obtain levosimendan ((yield—10.5 g, purity—99.5%).
..............................
one more patent
one more patent
U.S. Pat. No. 5,569,657 discloses the preparation of levosimendan and its salts. (±)-6-(4-aminophenyl)-5-methylpyridazin-3-(2H)-one is dissolved in 2-propanol on heating. L-tartaric acid is gradually added to the solution and stirred on heating to obtain a clear solution. The solution is cooled slowly to room temperature and then stirred overnight at 200 C to obtain a crystalline product. On filtering, the wet salt is dissolved in water and to it potassium carbonate solution is added with stirring. The free base obtained is filtered, washed with water and dried. The product is further dissolved in dioxane on heating and allowed to cool to room temperature. The contents are filtered and dried under vacuum to obtain (−)-6-(4-aminophenyl)-5-methylpyridazin-3-(2H)-one crystalline solid. The pure (−)-6-(4-aminophenyl)-5-methylpyridazin-3-(2H)-one compound is then treated with sodium nitrite and malononitrile under acidic condition to obtain levosimendan. A disadvantage of the resolution process disclosed is that to obtain a high optical purity (99.5%) of the pyridazinone compound, recrystallisation with dioxane is required. Also the process involves multiple steps and is time consuming.
(.+-.)-6-(4-aminophenyl)-5-methylpyridazin-3(2H)one (203 g, 1 mole) was dissolved in 2-propanol (40 dm.sup.3) on heating. To this solution (L)-tartaric acid (300 g, 2 mole) was gradually added. The mixture was stirred on heating until a clear solution was obtained. The solution was cooled slowly to room temperature with stirring. After it has been stirred over night at 20 The wet salt was dissolved in water (1.5 dm.sup.3) and potassium carbonate solution (190 g K.sub.2 CO.sub.3 in 0.75 dm.sup.3 of water) was added with stirring. The free base was filtered, washed with water and dried. The product (104.6 g) was dissolved in dioxane (0.6 dm.sup.3) on heating and allowed to cool to room temperature. The racemic 6-(4-aminophenyl)-5-methylpyridazin-3(2H)one was filtered (74.6 g) and the filtrate was evaporated to dryness in vacuo yielding (-)-6-(4-aminophenyl)-5-methylpyridazin-3(2H)one as a crystalline solid (23.8 g) with optical purity of 99.5%, m.p. 207 [a].sub.D.sup.25 =-383
(.+-.)-6-(4-aminophenyl)-5-methylpyridazin-3(2H)one (203 g, 1 mole) was dissolved in 2-propanol (10 dm.sup.3) on heating. To this solution (L)-tartaric acid (300 g, 2 mole) was gradually added. The mixture was stirred on heating until a clear solution was obtained and cooled slowly during 3 h to 50 C. The crystalline product was filtered and the procedure described in Example 1 was repeated. The yield of (-)-6-(4-aminophenyl)-5-methylpyridazin-3(2H)one was 30.3 g (97.4% of the theoretical). The optical purity was 99.7%. In total 140.8 g of the racemate was recovered.
The optical purities of the compounds were determined by the high performance liquid chromatography. The instrument was a Waters 600 E gradient pump with a Waters 991 photodiode array detector and a Waters 700 Satellite Wisp injector (Millipore Co.) controlled by a NEC Powermate SX Plus computer. The enantiomers of 6-(4-aminophenyl)-5-methylpyridazin-3(2H)one were separated by using a sellulose-type chiral column (Chiracel=OJ, 4.6.times.250 mm, Daicel Chemical Industries LTD.). The mobile phase consisted of 97% 2-propanol and 3% hexane. The flow rate was 0.3 ml/min. The enantiomers of [[4-(1,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl)phenyl]hydrazono]propanedinitrile were separated by using a β-cyclodextrin column (Cyclobond lb, 4.6.times.250 mm, Advance Separation Technologies Inc.). The mobile phase consisted of 41% methanol in water buffered to pH 4.0 with 1% triethylammonium acetate. The flow rate was 0.3 ml/min.
...............................
http://www.google.com/patents/WO2011007123
resolving agent used see patent
A process for preparing levosimendan substantially as herein described with reference to the examples. 19. A compound of formula Vl or a compound of formula VII substantially as herein described with reference to the examples.

...............................
http://www.google.com/patents/WO2011007123
resolving agent used see patent
A process for preparing levosimendan substantially as herein described with reference to the examples. 19. A compound of formula Vl or a compound of formula VII substantially as herein described with reference to the examples.

............................
resolution
http://www.google.com/patents/WO1992012135A1
The racemic 6-(4-amino- phenyl)-5-methyl-pyridazin-3(2H)one of formula (II)

can be synthesized by methods known in the literature (J. Med. Chem., 17, 273-281 (1974)). The resolution of the racemic compound (II) has, however, been proved very difficult because the 4-amino group in the molecule is weakly basic. The salts of 6-(4-amino-phenyl)-5-methylpyridazin-3(2H)one with optically active acids hydrolyse on crystallization readily back to the compound (II) and to the resolving compound which interfere the resolution procedure or make it totally impossible.
The separation of the pure enantiomers of compound (II) on a chiral HPLC-column has been described in European patent application EP 208518. This method is, however, not applicable for industrial scale. An enantio- selθctivθ seven step synthesis of (-)-6-(4-aminophenyl)-5-methylpyridazin- 3(2H)one starting from (+)-2-chloropropionic acid has also been described in the literature (J. Org.Chem., 56, 1963 (1991)). The total yield in this method is only 12 % giving (-)-6-(4-aminophenyl)-5-methylpyridazin-3(2H)one with an optical purity of 97.2 %.
It was now found that good enantiomeric separation of compound (II) could be obtained by using L- or D-tartaric acid in excess, preferably about 2 to about 3 equivalents, to the compound (II) in 2-propanol. The acid salts of (-)-6- (4-aminophenyl)-5-methylpyridazin-3(2H)one with L-tartaric acid 2-propanol solvate (Ilib) or corresponding (+)-6-(4-aminophenyl)-5-methylpyridazin- 3(2H)one with D-tartaric acid 2-propanol solvate (Ilia) crystallize in good yield and in practical optical purity.
(+) x L-tartaric acid x 2-propanol solvate J Ula
(-) x L-tartaric acid x 2-prσpanol solvate J nib

Example 6
Preparation of (-)-6-(4-aminophenyl)-5-methylpyridazin-3(2H)one by resolution of the corresponding racemate with L-tartaric acid.
(±)-6-(4-aminophenyl)-5-methylpyridazin-3(2H)one (203 g, 1 mole) was dissolved in 2-propanol (10 dm3) on heating. To this solution (L)-tartaric acid (300 g, 2 mole) was gradually added. The mixture was stirred on heating until a clear solution was obtained and cooled slowly during 3 h to 50°C and stirred further over night at 50°C. The crystalline product was filtered and the procedure described in Example 1 was repeated. The yield of (-)-6-(4-amino- phenyl)-5-methylpyridazin-3(2H)one was 30.3 g (97.4 % of the theoretical). The optical purity was 99.7 %. In total 140.8 g of the racemate was recovered.
The optical purities of the compounds were determined by the high performance liquid chromatography. The instrument was a Waters 600 E gradient pump with a Waters 991 photodiode array detector and a Waters 700 Satellite Wisp injector (Millipore Co.) controlled by a NEC Powermate SX Plus computer. The enantiomers of 6-(4-aminophenyl)-5-methylpyridazin-3(2H)one were separated by using a sellulose-type chiral column (Chiracel «OJ, 4.6x250 mm, Daicel Chemical Industries LTD.). The mobile phase consisted of 97 % 2- propanol and 3 % hexane. The flow rate was 0.3 ml/min. The enantiomers of [[4-(1 ,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl)phenyl]hydrazono]- propanedinitrile were separated by using a β-cyclodextrin column (Cyclobond lb, 4.6x250 mm, Advance Separation Technologies Inc.). The mobile phase consisted of 41 % methanol in water buffered to pH 4.0 with 1 % triethyl- ammonium acetate. The flow rate was 0.3 ml/min
................................
PATENTS
| US6355269 | 24 Sep 1998 | 12 Mar 2002 | Orion Corporation | Oral compositions of levosimendan |
| US6730673 | 8 Sep 2000 | 4 May 2004 | Orion Corporation | Pharmaceutical solutions of levosimendan |
| US6943164 | 10 Nov 2003 | 13 Sep 2005 | Orion Corporation | Pharmaceutical solutions of levosimendan |
| WO2011007123A1 | 12 Jul 2010 | 20 Jan 2011 | Cipla Limited | Process for preparing levosimendan and intermediates for use in the process |
| US6183771 * | Jun 26, 1997 | Feb 6, 2001 | Orion Corporation | Transdermal compositions containing levosimendan |
| EP0359439A1 | Aug 30, 1989 | Mar 21, 1990 | Orion-Yhtymä Oy | Substituted pyridazinones |
| EP0383449A2 | Jan 29, 1990 | Aug 22, 1990 | Orion-Yhtymä Oy | Pyridazinone derivatives and processes for preparing the same |
| GB383449A * | Title not available | |||
| GB565546A * | Title not available | |||
| WO1997035841A2 | Mar 27, 1997 | Oct 2, 1997 | Riikka Hyppoelae | Method for obtaining pure enantiomers of a pyridazinone derivative |
| WO2001000211A1 * | Jun 29, 2000 | Jan 4, 2001 | Lasse Lehtonen | A method for the treatment or prevention of coronary graft vasospasm |
| WO1993021921A1 * | May 5, 1993 | Nov 11, 1993 | Reijo Johannes Baeckstroem | Anti-ischemic medicament |
| WO1997035841A2 * | Mar 27, 1997 | Oct 2, 1997 | Riikka Hyppoelae | Method for obtaining pure enantiomers of a pyridazinone derivative |
| WO1999016443A1 * | Sep 24, 1998 | Apr 8, 1999 | Saila Antila | Oral compositions of levosimendan |
| WO1999032081A1 * | Dec 11, 1998 | Jul 1, 1999 | Marianne Karlsson | Transmucosal formulations of levosimendan |
| WO1999055305A2 * | Apr 23, 1999 | Nov 4, 1999 | Orion Corp | Controlled release peroral compositions of levosimendan |
| WO1999055337A1 * | Apr 23, 1999 | Nov 4, 1999 | Harjula Maarit | Stabile compositions comprising levosimendan and alginic acid |
| WO1999066912A2 * | Jun 18, 1999 | Dec 29, 1999 | Lasse Lehtonen | Method of treating pulmonary hypertension |
| WO2001000211A1 * | Jun 29, 2000 | Jan 4, 2001 | Lasse Lehtonen | A method for the treatment or prevention of coronary graft vasospasm |
| WO2002047603A2 * | Dec 14, 2001 | Jun 20, 2002 | Lasse Lehtonen | Use of levosimendan for treating erictile dysfunction |
| WO2003007962A1 * | Jul 4, 2002 | Jan 30, 2003 | Heimo Haikala | A combination therapy for the treatment of heart failure |
| WO2011007123A1 * | Jul 12, 2010 | Jan 20, 2011 | Cipla Limited | Process for preparing levosimendan and intermediates for use in the process |
| EP1612204A1 * | Mar 31, 2004 | Jan 4, 2006 | Daiichi Pharmaceutical Co., Ltd. | Hydrazone derivative |
| EP1619186A1 * | Mar 27, 1997 | Jan 25, 2006 | Orion Corporation | Method for obtaining pure enantiomers of a pyridazinone derivative |
| EP2143715A1 * | Mar 27, 1997 | Jan 13, 2010 | Orion Corporation | Method for obtaining pure enantiomers of a pyridazinone derivative |
| US5424428 * | Jan 3, 1992 | Jun 13, 1995 | Orion-Yhtyma Oy | (-)-[[4-(1,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl)phenyl]-hydrazono]pr |
| US5512571 * | May 31, 1995 | Apr 30, 1996 | Orion-Yhtyma Oy | (-) 4-(1,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl)phenyl!-hydra-zono!propanedinitrile |
| US5512572 * | May 5, 1993 | Apr 30, 1996 | Orion-Yhtyma Oy | Anti-ischemic medicament |
| US5569657 * | May 31, 1995 | Oct 29, 1996 | Orion-Yhtyma Oy | (-)-[[4-(1,4,5,6-tetrahydro-4-methyl-6-oxo-3-pyridazinyl)phenyl]-hydrazono]propanedinitrile |
| US5856480 * | Nov 10, 1995 | Jan 5, 1999 | Nippon Soda Co., Ltd. | Optically active compound |
| US6180789 | Mar 27, 1997 | Jan 30, 2001 | Orion Corporation | Method for obtaining pure enantiomers of a pyridazinone derivative |
| US6355269 | Sep 24, 1998 | Mar 12, 2002 | Orion Corporation | Oral compositions of levosimendan |
| US6399610 | Dec 11, 1998 | Jun 4, 2002 | Orion Corporation | Transmucosal formulations of levosimendan |
| US6462045 * | Jun 18, 1999 | Oct 8, 2002 | Orion Corporation | Method of treating pulmonary hypertension |
| US6531458 | Apr 23, 1999 | Mar 11, 2003 | Orion Corporation | Stable compositions comprising levosimendan and alginic acid |
| US6593329 | Jun 29, 2000 | Jul 15, 2003 | Orion Corporation | Method for the treatment or prevention of coronary graft vasospasm |
| US6949548 | Jul 4, 2002 | Sep 27, 2005 | Orion Corporation | Combination therapy for the treatment of heart failure |
| US7045147 | Apr 23, 1999 | May 16, 2006 | Orion Corporation | Controlled release peroral compositions of levosimendan |
| US7115604 | Dec 14, 2001 | Oct 3, 2006 | Orion Corporation | Method for treating erectile dysfunction |
| US7279479 | Feb 8, 2002 | Oct 9, 2007 | Orion Corporation | Method for the treatment of heart failure |
REFERENCES
Janssen et al., “Levosimendan improves diastolic and systolic function in failing human myocardium,” European Journal of Pharmacology, vol. 404, pp. 191-199 (2000).
Toivonen et al., “Electrophysiologic Effects of a Calcium Sensitizer Inotrope Levosimendan Administered Intravenously in Patients with Normal Cardiac Function,” Journal of Cardiovascular Pharmacology, vol. 35, pp. 664-669 (2000).
Alagona, ‘Advances in pacing for the patient with sick sinus syndrome’, Current Opinion in Cardiology, vol. 12, pp. 3-11, 1997.
Sundberg et al., ‘Hemodynamic and Neurohumoral Effects of Levosimendan, a New Calcium Sensitizer, at Rest and During Exercise in Healthy Men’, Am J Cardiol, vol. 75, pp. 1061-1066, 1995.
Lilleberg et al., ‘Dose-Range Study of a New Calcium Sensitizer, Levosimendan, in Patients with Left Ventricular Dysfunction’, Journal of Cardiovascular Pharmacology, vol. 26 (suppl. 1), pp. S63-S69, 1985.
Sandell et al., ‘Pharmacokinetics of Levosimendan in Healthy Volunteers and Patients with Congestive Heart Failure’, Journal of Cardiovascular Pharmacolohy, vol. 26, pp. 57-62, 1995.
Lilleberg et al., ‘Effects of a new calcium sensitizer, levosimendan, on haemodynamics, coronary blood flow and myocardial substrate utilization early after coronary artery bypass grafting’, European Heart Journal, vol. 19, pp. 660-668, 1998.
Harjola et al., ‘Oral Levosimendan Improves Cardiac Function and Hemodynamics in Patients with Severe Congestive Heart Failure’, Am J Cardiol, vol. 83, pp. 4 (l)-8 (l), 1999.
Singh et al., ‘Effects of Levosimendan on Cardiac arrhythimia : Electrophysiologic and Ambulatory Electrocardiographic Findings in Phase II and Phase III Clinical Studies in Cardiac Failure’, Am J Cardiol, vol. 83, pp. 16 (l)-20 (l), 1999.
Du Toit et al., ‘Levosimendan: Effects of a Calcium Sensitizer on Function and Arrhythmias and Cyclic Nucleotide Levels during Ischemia/Reperfusion in the Langendorff-Perfused Guinea Pig Heart’, The Journal of Pharmacology and Experimental Therapeutics, vol. 290, pp. 505-514, 1999.
......................................
INTERMEDIATES

| CAS Number: | 101328-85-2 |
| Molecular Weight: | 203.24 |
| Molecular Formula: | C11H13N3O |
| Chemical Name: | (R)-6-(4-Aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone |
| Synonyms: | OR-1855;(-)-SKF-93505;LEVOSIMENDAN DAZINONES INTERMEDIATES;(R)-6-(4-AMINOPHENYL)-5-METHYLPYRIDAZIN-3(2H)ONE;(-)-6-Aminophenyl-4,5-dihydro-5-methyl-3-(2H)pyridazinone;6-(4-AMINO-PHENYL)-5-METHYL-4,5-DIHYDRO-2H-PYRIDAZIN-3-ONE;(R)-6-(4-AMINOPHENYL)-4,5-DIHYDRO-5-METHYLPYRIDAZIN-3(2H)ONE;(r)-6-(4-aminophenyl)-4,5-dihydro-5-methyl-3(2h)-pyridazinone;(5R)-6-(4-Aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone;(R)-4,5-Dihydro-5-methyl-6-(4-aminophenyl)pyridazine-3(2H)-one |
.................................
PICKUP SYNTHESIS OF INTERMEDIATES
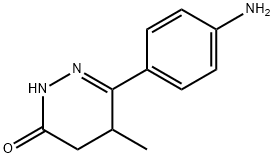 racemic
racemic
racemic| CAS No. | 36725-28-7 |
| Chemical Name: | 6-(4-Aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone |
| Synonyms: | ICI 109,081;LEVOSIMENDAN DAZINONES INTERMEDIATES;6-(4-Aminophenyl)-5-methylpyridazin-3(2H)one;6-(4-AMINOPHENYL)-5-METHYLPYRIDAZIN-3(2H)ONE, 98+%;6-(4-AMinophenyl)-4,5-dihydro-5-Methyl-3(2H)-pyrid;6-(4''-AMINOPHENYL)-4,5-DIHYDRO-5-METHYLPYRIDAZIN-3-ONE;6-(4-AMINOPHENYL)-4,5-DIHYDRO-5-METHYL-3(2H)PYRIDAZINONE;6-(p-Aminophenyl)-5-methyl-4,5-dihydro-3(2H)-pyridazinone;6-(p-Aminophenyl)-4,5-dihydro-5-methyl-3(2H)-pyridazinone;3-(4-aminophenyl)-4-methyl-4,5-dihydro-1H-pyridazin-6-one |
| Molecular Formula: | C11H13N3O |
| Formula Weight: | 203.24 |
6 - (4-aminophenyl)-5-methyl-4 ,5-dihydro-3 (2H)-pyridazinone is described in
J. Med. Chem., 17, 273-281 (1974)http://pubs.acs.org/doi/abs/10.1021/jm00249a004
SYNTHESIS OF
racemic compd
THIS IS ONLY STRUCTURE OF RACEMIC COMPD, IGNOrE THE WORD FORMULA II
BELOW IS A PART OF SYNTHESIS OF SOME OTHER COMPD, PLEASE PICK THIS COMPD OUT

Friedel-Crafts acylation of acetanilide (I) with propionyl chloride in the presence of AlCl3 afforded 4'-(acetamido) propiophenone (II). Subsequent Mannich reaction of (II) with dimethylamine hydrochloride and formaldehyde produced the aminoketone (III). Quaternization of the tertiary amine of (III) with MeI, followed by displacement of the resulting ammonium salt (IV) with KCN yielded the intermediate ketonitrile (V). In alternative procedure, nitrile (V) was prepared by acylation of acetanilide (I) with 3 -chloro-2-methylpropionyl chloride, and further displacement of the resulting beta-chloroketone (VI) with KCN. Acid hydrolysis of both nitrile and acetamido groups of (V) provided ketoacid (VII), which was cyclized with hydrazine to give pyridazinone ( VIII). Finally, acylation of the amino group of (VIII) by means of pyridine-2-carbonyl chloride furnished the title amide.
Liu, CM; et al;. Synthesis and platelet aggregation inhibitory activity of 6 - (4'-substituted acylaminophenyl) -4,5-dihydro-3-(2H)-pyridazinones Acta Pharm Sin 1999, 34, 1, 23.
ONE CAN EASILY CONSTRUCT SYNTHESIS FROM ABOVE SCHEME


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
 A new nanoparticle-based device can be used to monitor the quality of blood for transfusions
A new nanoparticle-based device can be used to monitor the quality of blood for transfusions







