Ann-Teresa Cusenza…..Managing Editor, Orphan Druganaut Blog « New Drug Approvals:
'via Blog this'
Friday, 14 March 2014
Monday, 10 March 2014
Umirolimus, Biolimus

Umirolimus, Biolimus
Biosensors (Originator)
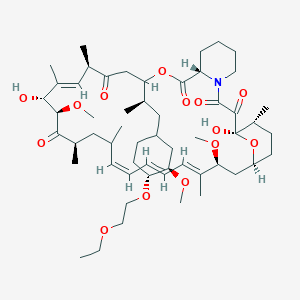
40 -O-[(2′-ethoxy) ethyl]rapamycin
(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-12-{(2R)-1-[(1S,3R,4R)-4-(2-Ethoxyethoxy)-3-methoxycyclohexyl]-2-propanyl}-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-11,36 -dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20-pentone
Umirolimus [INN], Umirolimus [USAN:INN], UNII-U36PGF65JH, TRM-986, 42-O-(2-ethoxyethyl) rapamycin, cas no 851536-75-9
Molecular Formula: C55H87NO14 Molecular Weight: 986.27758
Umirolimus (INN/USAN), is a macrocyclic lactone, a highly lipophilic derivative of sirolimus, an immunosuppressant. This drug is proprietary toBiosensors International, which uses it in its own drug-eluting stents, and licenses it to partners such as Terumo.
Biosensors had been developing a Biolimus A9(R)-eluting coronary stent for the treatment of arterial restenosis. No recent development has been reported. The product candidate was developed with BioMatrix(R), the company's low-profile, rapid-exchange delivery system. Specifically engineered for use on stents, Biolimus A9(R), a new rapamycin derivative, readily attaches to and enters smooth muscle cell membranes and binds to immunophilins inside the cell, causing cell cycle arrest at G0. Animal and in vitro studies suggest potency and safety profiles comparable to sirolimus.
Umirolimus inhibits T cell and smooth muscle cell proliferation, and was designed for use in drug eluting stents. This analog has a chemical modification at position 40 of the rapamycin ring. It has potent immunosuppressive properties that are similar to those of sirolimus, but the drug is more rapidly absorbed by the vessel wall, readily attaches and enters smooth muscle cell membranes causing cell cycle arrest at G0, and is comparable to sirolimus in terms of potency.
The key biologic event associated with the restenotic process is clearly the proliferation of smooth muscle cells in response to the expansion of a foreign body against the vessel wall. This proliferative response is initiated by the early expression of growth factors such as PDGF isoforms, bFGF, thrombin, which bind to cellular receptors.
However, the key to understanding the mechanism by which compounds like umirolimus inhibit cell proliferation is based on events which occur downstream of this growth factor binding. The signal transduction events which culminate in cell cycle arrest in the G1 phase are initiated as a result of ligand binding to an immunophilin known as FK binding protein-12. The FK designation was based on early studies conducted with tacrolimus, formerly known as FK-506, which binds this cytoplasmic protein with high affinity.
Subsequent investigations showed that rapamycin also binds to this intracellular target, forming an FKBP12–rapamycin complex which is not in itself inhibitory, but does have the capacity to block an integral protein kinase known as target of rapamycin (TOR). TOR was first discovered in yeast J.N. Heitman, N.R. Movva and M.N. Hall, Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast, Science 253 (1991), pp. 905–909. View Record in Scopus | Cited By in Scopus (434)and later identified in eukaryotic cells, where it was designated as mTOR, the mammalian target of rapamycin. The importance of mTOR is based on its ability to phosphorylate a number of key proteins, including those associated with protein synthesis (p70s6kinase) and initiation of translation (4E-BP1).
Of particular significance is the role that mTOR plays in the regulation of p27kip1, an inhibitor of cyclin-dependent kinases such as cdk2. The binding of agents like rapamycin and umirolimus to mTOR is thought to block mTOR's crucial role in these cellular events, resulting in arrest of the cell cycle, and ultimately, cell proliferation.
Introduction
It is known that Biolimus A9, a rapamycin derivative, is an immunosuppressant, and is also proven to have anti-tumor and anti-fungal effect.
Several prior arts had disclosed the improvements of the product yield of rapamycin derivatives. U.S. Pat. No. 7,193,078 to Isozaki et al. disclosed a process for producing Biolimus A9, giving an example to obtain a yield of 46% by reacting rapamycin with 2-ethoxyethyl trifluoromethane sulfonate (or 2-ethoxyethyl triflate) in an organic solvent.
However, the Isozaki's prior art still has the following drawbacks:
- 1. Even one example ever showed a 46% yield of Biolimus A9, it however just revealed a small-scale laboratory experiment with only one gram (1.09 mmol) of rapamycin and 1.95g (8.78 mmol) of 2-ethoxyethyl triflate. After amplifying or expanding the process to be larger scale, the yield will be remarkably reduced to thereby decrease the commercial or industrial value of this prior art (Note: The low yield after simulated process amplification will be hereinafter discussed in Examples 3, 4 of this application).
- 2. Even the reactant of 2-ethoxyethyl triflate is a compound with high activity, it is unstable and will be decomposed such as after being stored for one week at room temperature. Also, the triflate is not UV-absorbable and is therefore unsuitable for process tracking when proceeding the reaction. Such poor properties will affect the material storage, production scheduling and process tracking for commercially making the Biolimus A9.
sirolimus 42 - ether derivatives are a class of sirolimus derivative, is a new generation macrolide immunosuppressant and anticancer drugs. The compounds discovered by the Swiss company Sandoz, mainly applicable to organ transplant recipient's immune suppression and cancer. The synthesis of such substances currently on the patent literature have W09409010, CN102127092A and CN102268015A.
Patent Document W09409010 on Synthesis of this type of structure are used sirolimus protected materials in acidic or neutral reaction conditions, and then removing the protective group to obtain the target product. Such as 42-0 - (4 - hydroxymethyl) benzyl - sirolimus, the first synthesis of the formula V, and then removal of silyl ether protecting groups have the formula VI.

This synthetic method has several drawbacks: 1, the reaction reagent relatively difficult to obtain; 2, the intermediate prepared in the reaction yield is low;
3, sirolimus, structural part to participate in a two-step reaction, reduction reaction yield, costs.
CN102127092A mention a synthetic sirolimus 42 - ether derivative everolimus one way. This synthetic route similar to the W09409010 (route of reaction formula 1), but with silane reagents and reaction conditions are different.First reaction toluene as solvent, 50 ° C _60 ° C between the reaction and after-treatment of the intermediate after the first column chromatography, yield 32%.The second step in tetrahydrofuran as a solvent, the reaction overnight at 0 ° C, after treatment by a column chromatography to give the product, the yield was 66%, with a total yield of 21.1%.
Reaction Scheme I:

This method has the defects include: 1, the reaction reagent relatively difficult to obtain. 2, the structure part of sirolimus to attend two-step reaction, reduction reaction yield, costs. 3, the use of highly toxic solvents, are not suitable for practical application. 4, the reaction temperature is relatively harsh, difficult to control.
CN102268015A discloses a method for synthesizing everolimus. The first step to sirolimus or sirolimus derivatives as raw materials in -20 ° C was added dropwise trifluoromethanesulfonic anhydride and incubated for 3 hours, was isolated intermediates 02, the yield was 87.4% or 95.32%. The second step of the intermediate 02 with ethylene glycol mono-protected in 50 ° C reaction intermediate 03 was isolated in a yield of 79.0% or 76.78%. The third step of dilute hydrochloric acid was added dropwise at room temperature intermediate 03 was deprotected product everolimus. The total yield was 48.4% or 52.5%. See Reaction Scheme 2 synthetic route.
Reaction Scheme 2:

The method of the defects as follows: 1, to protection and deprotection of ethylene glycol. 2, the reaction steps excessive structural part to participate in the sirolimus-step reaction. 3, the reaction yield improved, but also greatly increased operating costs.
....................
.
Examples 5,42-0-(2_ ethoxy) ethyl - Synthesis of sirolimus
[0044] In the IOOml three-necked flask was added 2g of sirolimus, 2. 78g of 4_ dimethylaminopyridine, 5. 34g of chlorine acid glycol ester and 20ml of acetonitrile, 35 ° C The reaction was stirred 36 hours ended. The reaction solution was poured into an equal volume of saturated sodium bicarbonate solution and extracted with 5% potassium bisulfate solution was washed twice with a saturated sodium chloride solution, dried over anhydrous magnesium sulfate, filtered, and concentrated through the column. Silica gel column chromatography (EA: PE = I :20-2: 1), obtained by rotary evaporation 42-0 - (2 - ethoxy) ethyl - sirolimus I. 57g (yield: 73.3 %). HPLC analysis showed that: a purity of 88.2%.
............
A process for making Biolimus A9 represented by formula (1),

comprising reacting sirolimus of formula (2),

with
2-ethoxyethyl pentafluorobenzene sulfonate
under catalyzing by an organic base,
and in the presence of an organic solvent,
to undergo a nucleophilic substitution reaction to obtain Biolimus A9 of formula (1).
2. A process according to claim 1, wherein said 2-ethoxyethyl pentafluorobenzene sulfonate as used in the reaction is 1˜20 moles per mole of said sirolimus of formula (2).
the Biolimus A9 of the present invention will be presented as below-mentioned:
Biolimus A9
Biolimus A9

Reaction Parameters
Quantity of Alkylbenzene Sulfonate: 1˜20 equivalents, preferably being 5˜10 equivalents, per equivalent of sirolimus.
Reaction Temperature: 40˜80° C., preferably being 55˜65° C. Reaction Time: 12˜72 hours, preferably being 16˜30 hours.
After the reaction is completed, the rough product is collected, washed, dried and purified to obtain the Biolimus A9 of the present invention with high yield of 45%.
Since the product Biolimus A9 is a polyene macrolide, which is easily oxidized and decomposed during the storage or material handling.
Accordingly, a proper antioxidant may be homogeneously mixed with the Biolimus A9 to enhance the stability when stored or handled.
The proper antioxidants may be selected from: Butylatd hydroxytoluene (BHT), DL-α-tocopherol, propyl gallate, ascorbyl palmitate, 3-tert-butyl-4-hydroxyanisole, 2-tert-butyl-4-hydroxyanisole, and fumaric acid.
The Butylated hydroxytoluene (BHT) is the most preferable antioxidant adapted for use in the present invention.
The process for making Biolimus A9 in accordance with the present invention will be described in detail in view of the following examples:
EXAMPLE 1
A. Synthesis of 2-ethoxyethyl pentafluorobenzene sulfonate
In a reaction flask, 25 grams (93.8 mmol) of pentafluorobenzene sulfonyl chloride (or pentafluorobenzene sulfochloride) and 86 ml of tetrahydrofuran were added and nitrogen gas was filled into the flask.
The flask is then cooled to 0° C. and is dripped therein with 2-ethoxyethanol (8.5g, 94.5 mmol) and triethyl amine (15 g, 148.5 mmol). After dripping, the reaction solution is stirred for 30 minutes, and then filtered, concentrated and the residue is separated from the solution and further purified by silica gel column chromatography to obtain a colorless oily product of 2-ethoxyethyl pentafluorobenzene sulfonate (26.6 g, 83.1 mmol) having a yield of 88.6%.
B. Synthesis of Biolimus A9
In a reaction flask, 1 g (1.1 mmol) of sirolimus, 7.8 g (60.3 mmol) of ethyl di-isopropyl amine, 3.5 ml of methylene chloride and 2.8 g (8.7 mmol) of 2-ethoxyethyl pentrafluorobenzene sulfonate as previously obtained were added therein.
The reaction mixture in the flask was heated to 60° C. and agitated for 23 hours. It is cooled, and further added therein with ethyl acetate (100 ml) and aqueous solution of hydrochloric acid (1N, 100 ml ) under agitation.
Then, it is settled for separating the organic and aqueous layers. The organic layer is collected, and washed with pure water (100 ml) and saturated saline (100 ml). The washed organic liquid is then dried and concentrated. The residue is then separated from the liquid and further purified by silica gel column chromatography to obtain white solid product of Biolimus A9(0.49 g, 0.5 mmol) with a yield of 45.4%.
EXAMPLE 2 Process Amplification of Example 1B
In a reaction flask, 10 g (10.9 mmol) of sirolimus, 78 g (603.5 mmol) of ethyl di-isopropyl amine, 35 ml of methylene chloride and 28 g (87.4 mmol) of 2-ethoxyethyl pentrafluorobenzene sulfonate were added therein.
The reaction mixture in the flask was heated to 60° C. and agitated for 24 fours. It is cooled, and further added therein with ethyl acetate (500 ml) and aqueous solution of hydrochloric acid (1N, 500 ml ) under agitation.
Then, it is settled for separating the organic and aqueous layers. The organic layer is collected, and washed with pure water (500 ml) and saturated saline (400 ml). The washed organic liquid is then dried and concentrated. The residue is then separated from the liquid and further purified by silica gel column chromatography to obtain white solid product of Biolimus A9(4.8 g, 4.9 mmol) with a yield of 44.5%.
This example is a process amplification of the previous Example 1, Step B, by amplifying or expanding the quantity of each reactant for about 10 times of that of the Example 1 (of small scale).
By the way, the production yield (44.5%) of this Example is still as high as that of the previous Example 1 of small scale. It indicates that the reproducibility of high yield can still be obtained in accordance with the present invention even after process amplification, proving that the present invention is suitable for commercialization or mass production. The product may be further purified to obtain a high-purity final product of Biolimus A9 such as by middle-performance liquid chromatography or the like. The Biolimus A9 thus obtained is identified by the X-ray powder diffractogramm as shown in the single drawing FIGURE as attached herewith.
EXAMPLE 3 Comparative Example for Simulating the Process of the Prior Art of U.S. Pat. No. 7,193,078
A. Synthesis of 2-ethoxyethyl trifluoromethane sulfonate
In a reaction flask, 2-ethoxyethanol (10 g, 111 mmol), methylene chloride (177 ml) and 2,6-dimethyl pyridine (23.8 g, 222.3 mmol) were added into the flask, which is filled therein with nitrogen gas. It is cooled to 0° C. and added dropwise with trifluoromethane sulfonic acid anhydride (37.6 g, 133.4 mmol). After completing the dripping of said sulfonic acid anhydride, the reaction mixture is agitated for one hour and a saturated aqueous solution of ammonium chloride (20 ml) is added and further agitated for 10 minutes.
It is then settled for separating the layers. The organic layer is collected, and is respectively washed with aqueous solution of hydrochloric acid (1N, 100 ml), pure water (100 ml), saturated aqueous solution of sodium bicarbonate (100 ml) and saturated saline (100 ml). The washed organic layer is dried, concentrated and the residue is then separated and further purified with silica gel column chromatography to obtain the oily product of 22.5 g (101.3 mmol) of 2-ethoxyethyl trifluoromethane sulfonate (or 2-ethoxyethyl triflate), with a yield of 91.3%.
B. Synthesis of Biolimus A9
In a reaction flask, sirolimus (1 g, 1.1 mmol), ethyl di-isopropyl amine (7.8 g, 60.3 mmol), methylene chloride (3.5 ml) and 2-ethoxyethyl triflate (2.0 g, 8.8 mmol) as previously made in Example 3A were added into the flask, which is filled with nitrogen gas. The reaction mixture is heated to 60° C. and is agitated for one hour and twenty minutes. Then, it is cooled, added with ethyl acetate (100 ml) and aqueous solution of hydrochloric acid (1N, 100 ml) and is further agitated. After agitation, it is settled for separating the layers. The organic layer is collected and respectively washed with pure water (100 ml), saturated saline (80 ml). The washed organic layer is dried and concentrated. The residue is then separated and purified by silica gel column chromatography to obtain white product of Biolimus A9 (0.48 g, 0.49 mmol), with a yield of 44.5%.
EXAMPLE 4 Comparative Example for Simulative Process Amplification of Example 3B
In a reaction flask, sirolimus (10 g, 10.9 mmol), ethyl di-isopropyl amine (78 g, 603.5 mmol), methylene chloride (35 ml) and 2-ethoxyethyl triflate (20 g, 88 mmol), each having a quantity about 10 times of that used in Example 3B, were added into the flask, which is filled with nitrogen gas. The reaction mixture is heated to 60° C. and is agitated for one hour and twenty minutes. Then, it is cooled, added with ethyl acetate (500 ml) and aqueous solution of hydrochloric acid (1N, 500 ml) and is further agitated. After agitation, it is settled for separating the layers. The organic layer is collected and respectively washed with pure water (500 ml), saturated saline (400 ml). The washed organic layer is dried and concentrated. The residue is then separated and purified by silica gel column chromatography to obtain white product of Biolimus A9 (2.9 g, 2.9 mmol), having a yield of 26.8% only.
Comparatively, via this process amplification, the yield of Biolimus A9 of the prior art is remarkably reduced in comparison with its small-scale production (Example 3B). Therefore, the prior art of U.S. Pat. No. 7,193,078 may be considered as a process especially suitable for small-scale production, such as a laboratory experiment, rather than a large-scale commercial or industrial production, which is thus inferior to this application, when compared with this application which has shown the high yields both in small-scale process (Example 1) and large-scale process (Example 2).
Accordingly, this application is more suitable for commercialization for mass production.
Moreover, the essential reactant of 2-ethoxyethyl triflate of the prior art (U.S. Pat. No. 7,193,078), even having high activity, is unstable because it will be decomposed into unknown compounds after one-week storage (by NMR spectrographic detection) as accompanied with physical change from its original colorless transparent liquid to a black viscous oily product, to thereby be inferior to this application because the 2-ethoxyethyl pentafluoro benzene sulfonate (which is obviously different from the 2-ethoxyethyl triflate as used in the prior art) of this application is still stable after one-week storage as aforementioned.
Furthermore, the 2-ethoxyethyl pentafluorobenzene sulfonate of this application may absorb ultra-violet rays to have a better tractability during the process proceeding than that of the 2-ethoxyethyl triflate (which is not UV-absorbable) of the prior art. So, this application is also beneficial for better production scheduling, reliable process tracking and efficient production management than the prior art.
So, this application is more suitable for commercial production even when considering the stability of product storage and improvement of process monitoring, control and management.
EXAMPLE 5
The Biolimus A9, as obtained from Example 2, is respectively added with anti-oxidant, namely Butylated Hydroxytoluene (or BHT), for 0.1%, 0.2, 0.5%, and 1% (w/w) based on 100% (wt) of Biolimus to enhance its stability at 40° C. by revealing a high yield of more than 99.4% even after six-week storage. Comparatively, a control test is provided by adding 0% of anti-oxidant (BHT) into Biolimus A9, resulting in a reduction of yield to be 69.7% after six-week storage. The yield data of different amounts of anti-oxidant as added into Biolimus A9 with respect to time lapse of weeks are summarized in Table 1 as below-mentioned.
...............................
The O-(2-ethoxyethyl)-rapamycin can be produced by reaction between rapamycin and 2-ethoxyethyl triflate in the presence of N,N-diisopropylethylamine in methylene chloride.

An example of the O-alkylrapamycin derivative (with R=hydroxyalkyl) is O-(2-hydroxyethyl)-rapamycin represented by the general formula 3 below.
The O-(2-hydroxyethyl)-rapamycin can be produced by reaction between rapamycin and t-butyldimethylsilyloxyethyl triflate in the presence of N,N-diisopropylethylamine in methylene chloride, followed by deprotecting of t-butyldimethylsilyl group.

EXAMPLES
The invention will be described with reference to the following examples, which demonstrate the efficient production of O-alkylrapamycin derivatives by the process of the present invention.
Example 1
- (1) Synthesis of 2-ethoxyethyl Triflate
In a round bottom flask containing a stirring bar was placed 9.0 g (100 mmol) of ethoxyethanol. The atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. The flask was given 160 mL of methylene chloride and 23.3 mL (120 mmol) of 2,6-lutidine. The flask cooled with ice was given dropwise 20.2 mL (120 mmol) of trifluoromethanesulfonic acid anhydride over 20 minutes. After stirring for 1 hour, the reaction liquid was mixed with 20 mL of saturated solution of ammonium chloride. The resulting mixture was washed sequentially with 1N hydrochloric acid (100 mL), deionized water (100 mL), saturated solution of sodium hydrogen carbonate (100 mL), and saturated aqueous solution of sodium chloride (100 mL). The organic layer was separated and dried with anhydrous sodium sulfate. With the sodium sulfate filtered off, the solution was concentrated under reduced pressure. The residue underwent silica gel chromatography. Thus there was obtained 15.03 g (67.6% yields) of 2-ethoxyethyl triflate from the fraction in eluate of 20% ethyl acetate-hexane.
- (2) Synthesis of 40-O-[(2′-ethoxy)ethyl]rapamycin
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of methylene chloride for dissolution. To the flask was further added 10 mL (57.5 mmol) of N,N-diisopropylethylamine and 1.95 g (8.78 mmol) of the previously synthesized 2-ethoxyethyl triflate with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated by using a rotary evaporator. The concentrated solution was purified using a column chromatograph, with a silica gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1 v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 494 mg (0.501 mmol) of the desired product (46% yields).
Example 2
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of chloroform for dissolution. To the flask was further added 10 mL (57.5 mmol) of N,N-diisopropylethylamine and 1.95 g (8.78 mmol) of the 2-ethoxyethyl triflate previously synthesized in Example 1 with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated using a rotary evaporator. The concentrated solution was purified using column chromatograph, with a silica gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1, v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 451 mg (0.458 mmol) of the desired product (42% yields).
Example 3
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of methylene chloride for dissolution. To the flask was further added 8 mL (57.4 mmol) of triethylamine and 1.95 g (8.78 mmol) of the 2-ethoxyethyl triflate previously synthesized in Example 1 with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated using a rotary evaporator. The concentrated solution was purified using column chromatograph, with a silica-gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1, v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 344 mg (0.349 mmol) of the desired product (32% yields).
Example 4
In 2 mL of methanol was dissolved 500 mg of the 40 -O-[(2′-ethoxy)ethyl]rapamycin which had been obtained in Example 1. The resulting solution was added dropwise to 20 mL of deionized water with stirring. The solids which had precipitated out were filtered off and washed with a small amount of water and finally dried under reduced pressure at 40° C. for more than 10 hours. Thus there was obtained 483 mg of white powder.
This product gave an NMR chart as shown in FIG. 1. This NMR chart indicates the structure of 40 -O-[(2′-ethoxy) ethyl]rapamycin represented by the general formula 4.

Comparative Example
A sample of 40-O-[(2′-ethoxy)ethyl]rapamycin was synthesized by the process disclosed in WO94/09010 official gazette so as to evaluate yields.
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of toluene for dissolution. To the flask was further added 467 mg (4.36 mmol) of 2,6-lutidine and 1.95 g (8.78 mmol) of the 2-ethoxyethyl triflate previously synthesized in Example 1 with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated using a rotary evaporator. The concentrated solution was purified using column chromatograph, with a silica gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1, v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 247 mg (0.251 mmol) of the desired product (23% yields).
NMR

This product gave an NMR chart as shown in FIG. 1. This NMR chart indicates the structure of 40-O-[(2′-ethoxy)ethyl]rapamycin represented by the general formula 4.

| US20050101624 | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20050131008 | Nov 12, 2004 | Jun 16, 2005 | Sun Biomedical, Ltd. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US7193078 * | Mar 1, 2005 | Mar 20, 2007 | Terumo Kabushiki Kaisha | Process for production of O-alkylated rapamycin derivatives |
| WO2012017449A1 | Aug 2, 2011 | Feb 9, 2012 | Meril Life Sciences Pvt. Ltd | Process for preparation of novel 42-0-(heteroalkoxyalkyl) rapamycin compounds with anti-proliferative properties" |
| US7872122 | May 8, 2009 | Jan 18, 2011 | Chunghwa Chemical Synthesis & Biotech Co., Ltd. | Process for making Biolimus A9 |
Saturday, 8 March 2014
Friday, 7 March 2014
Subscribe to:
Posts (Atom)