Bitter Apricot Seed ( Xingren ) 杏仁 « New Drug Approvals:
'via Blog this'
Saturday, 29 March 2014
DS-8587 (Daiichi Sankyo (Japan) a new broad-spectrum antibacterial agent, is in phase I clinical trials for the treatment of bacterial infection.

DS-8587
Daiichi Sankyo (Japan)
7-[3a(R)-Amino-6a(S)-fluoroperhydrocyclopenta[c]pyrrol-2-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropyl]-8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride dihydrate
7-[(1S,6S)-1-amino-4-oxa-8-azabicyclo[4.3.0]nonan-8-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropan-1-yl]-1,4-dihydro-8-methoxy-4-oxoquinoline-3-carboxylic acid
| C21 H22 F3 N3 O3 . Cl H . 2 H2 O | |
| Mw | 493.904 |
DS-8587, a new broad-spectrum antibacterial agent, is in phase I clinical trials at Daiichi Sankyo for the treatment of bacterial infection.
DS-8587, from Daiichi Sankyo, is a fluoroquinolone with improved activity against both Gram-negative and Gram-positive bacteria. The compound is especially effective against Acinetobacter baumannii but also has improved activity against streptococci, staphylococci, enterococci, E. coli, and anaerobes . The compound is currently under Phase I of clinical development .
DS-8587, a new generation of fluoroquinolone, against Acinetobacter baumannii. The MICs against clinical isolates and inhibitory activity against target enzymes of DS-8587 was superior to ciprofloxacin and levofloxacin. Furthermore, the antibacterial activity of DS-8587 was less affected by adeA/adeB/adeC or abeM efflux pumps and frequency of single-step mutations with DS-8587 was lower as compared to those with ciprofloxacin. DS-8587 might be an effective agent against A. baumanniiinfection.
WO 2008082009 or
http://www.google.com/patents/EP2540715A1?cl=en
- [Reference Example 71]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester


- [(3S)-3-(3-Hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (46 g) and imidazole (11.9 g) were dissolved in dimethylformamide (600 mL). After addition of tert-butyldimethylsilyl chloride (23.2 g) under ice-cooling, the mixture was stirred at room temperature for 59.5 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was sequentially washed with saturated sodium bicarbonate water and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2 -> 2:1) to give 29.7 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.22 (5H, m), 5.48 (1H, q, J=7.11 Hz), 3.58 (2H, t, J=6.13 Hz), 3.34 (1H, d, J=10.05 Hz), 3.12 (1H, d, J=10.05 Hz), 2.94 (1H, d, J=16.91 Hz), 2.31 (1H, d, J=17.16 Hz), 1.86-1.74 (1H, m), 1.72-1.62 (1H, m), 1.51 (3H, d, J=7.11 Hz), 1.49-1.24 (2H, m), 1.33 (9H, s), 0.88 (9H, s), 0.03 (6H, s).
[Reference Example 72]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

- (3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (30 g) was dissolved in tetrahydrofuran (280 mL), and the atmosphere was replaced with argon. Then, lithium hexamethyldisilazide (1.0 M solution in tetrahydrofuran) (78.0 mL) was added dropwise at -15°C, and the mixture was stirred at -5°C for 30 minutes. After cooling to -15°C again, a solution of N-fluorobenzenesulfonimide (26.6 g) in tetrahydrofuran (220 mL) was added dropwise, and the mixture was stirred at room temperature for 17 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2) to give 8.15 g of the title compound as a pale yellow solid. 1H-NMR (400 MHz, CDCl3) δ: 7.37-7.23 (5H, m), 5.53-5.44 (1H, m), 5.18 (1H, d, J=51.72 Hz), 3.64-3.52 (2H, m), 3.32-3.19 (2H, m), 1.92-1.65 (2H, m), 1.55 (3H, d, J=4.66 Hz), 1.33 (9H, s), 0.88 (9H, s), 0.03 (6H, s).
MS (FAB) m/z: 480 (M+H)+.
IR (ATR) ν: 3421, 2977, 2935, 2877, 1698, 1454, 1369, 1309, 1249, 1153, 1058, 1035, 1006, 842 cm-1.
[Reference Example 73]
(3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

- (3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (8.15 g) was dissolved in tetrahydrofuran (25.0 mL). Acetic acid (22.0 mL) and tetrabutylammonium fluoride (1.0 M solution in tetrahydrofuran) (25.0 mL) were added under ice-cooling, and the mixture was stirred at room temperature for 21.5 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was sequentially washed with saturated sodium bicarbonate water and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2 -> 1:1) to give 5.77 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.22 (5H, m), 5.48 (1H, q, J=7.03 Hz), 5.20 (1H, d, J=51.48 Hz), 3.69-3.59 (2H, m), 3.31-3.21 (2H, m), 1.95-1.72 (2H, m), 1.68-1.43 (2H, m), 1.56 (3H, d, J=7.11 Hz), 1.33 (9H, s).
[Reference Example 74]
(3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

- (3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (12.20 g) was dissolved in dichloromethane (400 mL). Benzenesulfonyl chloride (9.06 mL), triethylamine (10.7 mL), and 4-dimethylaminopyridine (2.04 g) were added under ice-cooling, and the mixture was stirred at room temperature for 12.5 hours. Saturated sodium bicarbonate water was added to the reaction solution, and the mixture was stirred for 30 minutes, followed by extraction with dichloromethane. The organic layer was sequentially washed with a 10% citric acid solution and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 8:2 -> 1:1) to give 11.7 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.94 - 7.87 (2H, m), 7.71-7.63 (1H, m), 7.60-7.53 (2H, m), 7.37-7.23 (5H, m), 5.46 (1H, q, J=7.11 Hz), 5.15 (1H, d, J=51.48 Hz), 4.10-3.98 (2H, m), 3.26-3.15 (2H, m), 1.88-1.50 (4H, m), 1.55 (3H, s), 1.30 (9H, s).
[Reference Example 75]
(1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester

- (3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (10.9 g) was dissolved in tetrahydrofuran (350 mL), and the atmosphere was replaced with argon. Then, potassium hexamethyldisilazide (0.5 M solution in toluene) (86.5 mL) was added dropwise at - 15°C, and the mixture was stirred at 0°C for 1.5 hours. After cooling to -10°C, saturated aqueous ammonium chloride (100 mL) was added dropwise, and the mixture was stirred at room temperature for 30 minutes. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 7:1) to give 4.36 g of the title compound as a pale yellow solid.
1H-NMR (400 MHz, CDCl3) δ: 7.38-7.25 (5H, m), 5.58-5.49 (1H, m), 3.63 (1H, d, J=10.3 Hz), 2.91 (1H, dd, J=10.3, 3.2 Hz), 2.67-2.56 (1H, m), 2.50-2.38 (1H, m), 2.26-2.09 (1H, m), 2.06-1.94 (1H, m), 1.74-1.66 (1H, m), 1.54 (3H, d, J=7.1 Hz), 1.50-1.40 (1H, m), 1.34 (9H, s).
[Reference Example 76]
(1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane


- (1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester (4.36 g, 12.5 mmol) was dissolved in dichloromethane (70 mL). Trifluoroacetic acid (70 mL) was added dropwise, and the mixture was stirred at room temperature for six hours. The solvent was evaporated under reduced pressure, and then the residue was azeotropically distilled with toluene to give carboxylic acid (3.70 g).
- The resulting carboxylic acid was dissolved in toluene. Triethylamine (3.51 mL, 25.2 mmol) and diphenylphosphoryl azide (2.98 ml, 13.8 mmol) were added, and the mixture was heated to reflux for five hours. The solvent was evaporated under reduced pressure. Then, 1,4-dioxane (110 ml) and 6N hydrochloric acid (110 mL) were added to the residue, and the mixture was stirred at 60°C for 2.5 hours. After extraction with water and ethyl acetate, the aqueous layer was made alkaline with a saturated sodium hydroxide solution and extracted with chloroform twice. The organic layers were combined, dried over anhydrous sodium sulfate, and filtered, and then the solvent was evaporated under reduced pressure. Di-tert-butyl dicarbonate (11.05 g) was added to the residue, and the mixture was stirred at 75°C for six hours. The reaction solution was concentrated under reduced pressure, and then the residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 1:1) to give 3.69 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.23 (5H, m), 5.50 (1H, q, J=7.1 Hz), 5.22 (1H, brs), 3.34 (2H, s), 2.49-2.37 (1H, m), 2.32-2.03 (3H, m), 2.02-1.90 (1H, m), 1.51 (3H, d, J=7.1 Hz), 1.55-1.48 (1H, m), 1.35 (9H, s).
[Reference Example 77]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane

- (1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane (3.69 g, 10.2 mmol) was dissolved in tetrahydrofuran (200 mL). A 1.20 M solution of a borane-tetrahydrofuran complex in tetrahydrofuran (42.4 mL, 50.9 mmol) was added dropwise under ice-cooling, and the mixture was stirred for two hours while gradually heating to room temperature. The solvent was evaporated under reduced pressure. Under ice-cooling, 90% aqueous ethanol (100 mL) and triethylamine (100 mL) were added to the residue, and the mixture was heated to reflux for two hours. The solvent was evaporated under reduced pressure, and then the residue was extracted with saturated sodium bicarbonate water and dichloromethane. Thereafter, the target substance was extracted from the aqueous layer with dichloromethane. The organic layers were combined, washed with brine, and dried over anhydrous sodium sulfate. After filtration, the solvent was evaporated under reduced pressure, and the resulting residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 95:5 -> 90:10) to give 3.33 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.32 - 7.18 (5H, m), 5.38 (1H, brs), 3.22 (1H, q, J=6.37 Hz), 2.92-2.57 (4H, m), 2.12-1.86 (4H, m), 1.80-1.67 (1H, m), 1.63-1.52 (3H, m), 1.42 (9H, s), 1.32 (3H, d, J=6.37 Hz)
- [Reference Example 78]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane

- (1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane (700 mg, 2.0 mmol) was dissolved in ethanol (30 mL). 10% palladium-carbon (50% wet) (1.01 g) was added, and the mixture was stirred in a hydrogen atmosphere at 50°C for 15 hours. The catalyst was removed by filtration, and then the filtrate was concentrated under reduced pressure. The resulting residue was subjected to silica gel column chromatography (dichloromethane:methanol = 98:2 -> 95:5) to give 446 mg of the title compound as a pale yellow solid.
[α]D 23-15° (c=0.100, MeOH).
1H-NMR (400 MHz, CDCl3) δ: 5.29 (1H, brs), 3.47-3.18 (2H, m), 2.93-2.79 (2H, m), 2.15-1.71 (6H, m), 1.45 (9H, s).
- [Example 17]
7-[(1R,5S)-1-Amino-5-fluoro-3-azabicyclo[3.3.0]octan-3-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid

- Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were added to a solution of (1R,5S)-1-(tert-butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane (250 mg, 1.02 mmol) in dimethyl sulfoxide (5.0 mL). The mixture was stirred at room temperature for seven days. Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were further added to the reaction solution, and the mixture was stirred at room temperature for seven days. Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were further added to the reaction solution, and the mixture was stirred at room temperature for ten days. Ethanol (6.0 mL), water (2.0 mL), and triethylamine (2.0 mL) were added to the reaction solution, and the mixture was stirred at 80°C for one hour. The solvent was evaporated under reduced pressure, and then the residue was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with water twice and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (dichloromethane:methanol = 98:2), and the resulting fraction was concentrated under reduced pressure. Then, the residue was dissolved in concentrated hydrochloric acid (3.5 mL) under ice-cooling, and the solution was stirred at room temperature for one hour. The reaction solution was washed with chloroform five times, and then the aqueous layer was adjusted to pH 12 with a saturated sodium hydroxide solution. The basic solution was adjusted to pH 7.4 with hydrochloric acid, followed by extraction with chloroform. The organic layer was dried over anhydrous sodium sulfate and filtered, and then the solvent was evaporated under reduced pressure. The resulting residue was purified by PTLC (developed with the lower layer of chloroform:methanol:water = 7:3:1). The resulting residue was solidified with isopropanol to give 7.1 mg of the title compound as a pale yellow solid.
- 1H-NMR (400 MHz, 0.1N NaOD) δ: 8.50 (1H, s), 7.77 (1H, d, J=13.73 Hz), 5.80-4.80 (1H, m), 4.22-4.10 (1H, m), 3.99-3.85 (1H, m), 3.68-3.47 (2H, m), 3.43-3.34 (1H, m), 2.68 (3H, s), 2.21-1.98 (3H, m), 1.97-1.56 (4H, m), 1.42-1.23 (1H, m).
MS (FAB); m/z: 422 (M+H)+.
Anal.Calcd C21H22F3N3O3·0.5H2O·0.25IPA: C, 58.65; H, 5.66; F, 12.80; N, 9.43. Found: C, 58.63; H, 5.35; F, 12.35; N, 9.22.
IR (ATR) ν: 2971, 2856, 1722, 1614, 1450, 1432, 1322, 1132, 1097, 987, 954, 929, 887, 856, 804 cm-1.
WO 2012018105
http://www.google.st/patents/WO2012018105A1?cl=en
The following structural formula

compound represented by, 7 - [(1R, 5S) -1 - amino-5 - fluoro-3 - azabicyclo [3.3.0] octan-3 - yl] -6 - fluoro -1 - [(1R, . the 2S) -2 - fluoro-cyclopropane-1 - yl] -1,4 - dihydro-8 - methyl-4 - oxo-3-quinoline -) is referred to as carboxylic acid (hereinafter referred to as Compound A multi-agent containing quinolone resistance including resistant Gram-positive cocci resistant pneumococcus, etc., widely against gram-negative bacteria from Gram-positive bacteria, and, in addition to show strong antibacterial activity, convulsions, which is known in the art as a side effect of the antimicrobial agent of the present system potential cardiotoxicity light and toxicity-inducing activity (photosensitivity), has been reported recently in clinical further (QT prolongation), blood sugar abnormalities, and to express the side effects of delayed-type drug 疹等 is excellent safety low, Then, it is excellent oral absorbability and organ migration properties become apparent, is expected as an antimicrobial agent superior (Patent Document 1).
WO 2008/082009 pamphlet
The compound A, was synthesized according to the method described in Patent Document 1.
Preparation 7 1 hydrochloride dihydrate Ratings (1) Compound A crystalline acid addition salt preparation of acid addition salts of (Example 1) Compound A, and Compound A - [(1R, 5S) - 1 - amino-5 - fluoro-3 - azabicyclo [3.3.0] octan-3 - yl] -6 - fluoro -1 - [(1R, 2S) -2 - fluoro-cyclo-1 - yl] -1 , 4 - dihydro-8 - methyl-4 - oxo-3-quinoline - was added 1mol / L hydrochloric acid (74μL) carboxylic acid (Compound A) (31.3mg,, 0.074mmol) in, and dried under reduced pressure at room temperature.10% aqueous 2 the residue - was added to (100μL) propanol was dissolved by heating at 60 ℃, and allowed to stand day out on the room temperature. Collected by filtration the precipitated crystals, and the 1st air dried, 19.9mg (yield: 54%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 2H 2 O
Theoretical value: C; 51.07, H; 5.51, N; 8.51, F; 11.54, Cl; 7.18
Measured value: C; 50.93, H; 5.40, N; 8.49, F; 11.30, Cl; 7.47
Preparation 7 1 hydrochloride dihydrate Ratings (1) Compound A crystalline acid addition salt preparation of acid addition salts of (Example 1) Compound A, and Compound A - [(1R, 5S) - 1 - amino-5 - fluoro-3 - azabicyclo [3.3.0] octan-3 - yl] -6 - fluoro -1 - [(1R, 2S) -2 - fluoro-cyclo-1 - yl] -1 , 4 - dihydro-8 - methyl-4 - oxo-3-quinoline - was added 1mol / L hydrochloric acid (74μL) carboxylic acid (Compound A) (31.3mg,, 0.074mmol) in, and dried under reduced pressure at room temperature.10% aqueous 2 the residue - was added to (100μL) propanol was dissolved by heating at 60 ℃, and allowed to stand day out on the room temperature. Collected by filtration the precipitated crystals, and the 1st air dried, 19.9mg (yield: 54%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 2H 2 O
Theoretical value: C; 51.07, H; 5.51, N; 8.51, F; 11.54, Cl; 7.18
Measured value: C; 50.93, H; 5.40, N; 8.49, F; 11.30, Cl; 7.47
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 5.3,7.9,10.6,13.3,21.1,23.0,25.1,27.6 (°)
2 5% aqueous (1001.6mg, 0.746mmol) in the preparation of Compound A one hydrochloride monohydrate (2) Compound A - was added propanol (30mL), was dissolved by heating at 60 ℃. After stirring day out on the room temperature and stirred for 6 hours at 10 ℃. Collected by filtration the precipitated crystals, and the 1st dried air, 839.3 mg (yield: 87%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 1H 2 O
Theoretical value: C; 53.00, H; 5.30, N; 8.83, F; 11.98, Cl; 7.45
Measured value: C; 53.25, H; 5.43, N; 8.51, F; 11.58, Cl; 7.18
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 11.3,14.0,20.1,21.4,22.8,24.0,26.0,26.6 (°)
2 5% aqueous (1001.6mg, 0.746mmol) in the preparation of Compound A one hydrochloride monohydrate (2) Compound A - was added propanol (30mL), was dissolved by heating at 60 ℃. After stirring day out on the room temperature and stirred for 6 hours at 10 ℃. Collected by filtration the precipitated crystals, and the 1st dried air, 839.3 mg (yield: 87%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 1H 2 O
Theoretical value: C; 53.00, H; 5.30, N; 8.83, F; 11.98, Cl; 7.45
Measured value: C; 53.25, H; 5.43, N; 8.51, F; 11.58, Cl; 7.18
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 11.3,14.0,20.1,21.4,22.8,24.0,26.0,26.6 (°)
ref
- Higuchi, S.; Onodera, Y.; Chiba, M.; Hoshino, K.; Gotoh, N. Potent in vitro antibacterial activity of DS-8587, a new generation of broad spectrum quinolone, against Acinetobacter baumannii. Antimicrob. Agents Chemother. 2013, doi:10.1128/AAC.02374-12.
- Daiichi Sankyo. Major R&D pipeline as of July, 2013. Available online: http://www.daiichisankyo.com/rd/pipeline/pdf/Pipeline_EN.pdf (accessed on 28 September 2013).
| EP0343524A1 | May 19, 1989 | Nov 29, 1989 | Shionogi Seiyaku Kabushiki Kaisha | Pyridonecarboxylic acids and antibacterial agents |
| JPH0395176A | Title not available | |||
| JPH02231475A | Title not available | |||
| JPH08225567A | Title not available | |||
| JPS6345261A | Title not available | |||
| JPS6456673A | Title not available | |||
| JPS61282382A | Title not available | |||
| US5017708 * | Sep 8, 1989 | May 21, 1991 | Shionogi & Co., Ltd. | Azabicycloalkanes |
| WO1994014794A1 | Dec 28, 1993 | Jul 7, 1994 | Hideki Ao | 8-methoxyquinolonecarboxylic acid derivative |
| WO1995021163A1 | Feb 2, 1995 | Aug 10, 1995 | Katsumi Chiba | Pyridonecarboxylic acid derivative substituted by bicyclic amino group, ester thereof, salt thereof, and bicyclic amine as intermediate therefor |
| WO1996023782A1 | Feb 1, 1996 | Aug 8, 1996 | Daiichi Seiyaku Co | Heterocyclic compounds |

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.
Friday, 28 March 2014
New Drug Approvals « All about Drugs, live, by DR ANTHONY MELVIN CRASTO, Worlddrugtracker, Helping millions, 6 million hits on google, pushing boundaries, one lakh plus connections worldwide, 191000 plus VIEWS on this blog in 179 countries
Thursday, 27 March 2014
Aldoxorubicin…….Treatment of cancer …HIV-derived Kaposi’s Sarcoma, pancreatic cancer and for the treatment of soft tissue sarcoma.


Aldoxorubicin
http://www.ama-assn.org/resources/doc/usan/aldoxorubicin.pdf
 in phase 3
in phase 3
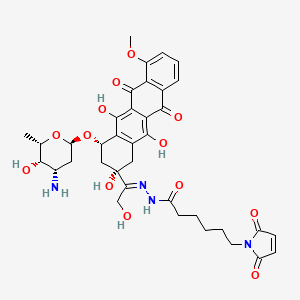
(E)-N'-(1-((2S,4S)-4-(((2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-2-yl)-2-hydroxyethylidene)-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanehydrazide hydrochloride
1H-Pyrrole-1-hexanoic acid, 2,5-dihydro-2,5-dioxo-, (2E)-2-[1-[(2S,4S)-4-[(3-amino-
2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-1,2,3,4,6,11-hexahydro-2,5,12-trihydroxy-
7-methoxy-6,11-dioxo-2-naphthacenyl]-2-hydroxyethylidene]hydrazide
2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-1,2,3,4,6,11-hexahydro-2,5,12-trihydroxy-
7-methoxy-6,11-dioxo-2-naphthacenyl]-2-hydroxyethylidene]hydrazide
N'-[(1E)-1-{(2S,4S)-4-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-2,5,12-
trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-2-yl}-2-
hydroxyethylidene]-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanohydrazide
MOLECULAR FORMULA C37H42N4O13
trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-2-yl}-2-
hydroxyethylidene]-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanohydrazide
MOLECULAR FORMULA C37H42N4O13
MOLECULAR WEIGHT 750.7
SPONSOR CytRx Corp.
CODE DESIGNATION INNO-206
CAS REGISTRY NUMBER 1361644-26-9
CAS: 151038-96-9 (INNO-206); 480998-12-7 (INNO-206 HCl salt), 1361644-26-9

hydrochloride
CAS: 151038-96-9
Chemical Formula: C37H42N4O13
Exact Mass: 750.27484
Molecular Weight: 750.75
Certificate of Analysis:
| |
QC data:
| |
Safety Data Sheet (MSDS):
|

In vitro protocol:
|
Clin Cancer Res. 2012 Jul 15;18(14):3856-67
|
In vivo protocol:
|
Clin Cancer Res. 2012 Jul 15;18(14):3856-67.
Invest New Drugs. 2010 Feb;28(1):14-9.
Invest New Drugs. 2012 Aug;30(4):1743-9.
Int J Cancer. 2007 Feb 15;120(4):927-34.
|
Clinical study:
|
Expert Opin Investig Drugs. 2007 Jun;16(6):855-66.
|

Aldoxorubicin (INNO-206): Aldoxorubicin, also known as INNO-206, is the 6-maleimidocaproyl hydrazone derivative prodrug of the anthracycline antibiotic doxorubicin (DOXO-EMCH) with antineoplastic activity. Following intravenous administration, doxorubicin prodrug INNO-206 binds selectively to the cysteine-34 position of albumin via its maleimide moiety. Doxorubicin is released from the albumin carrier after cleavage of the acid-sensitive hydrazone linker within the acidic environment of tumors and, once located intracellularly, intercalates DNA, inhibits DNA synthesis, and induces apoptosis. Albumin tends to accumulate in solid tumors as a result of high metabolic turnover, rapid angiogenesis, hyervasculature, and impaired lymphatic drainage. Because of passive accumulation within tumors, this agent may improve the therapeutic effects of doxorubicin while minimizing systemic toxicity.
“Aldoxorubicin has demonstrated effectiveness against a range of tumors in both human and animal studies, thus we are optimistic in regard to a potential treatment for Kaposi’s sarcoma. The current standard-of-care for severe dermatological and systemic KS is liposomal doxorubicin (Doxil®). However, many patients exhibit minimal to no clinical response to this agent, and that drug has significant toxicity and manufacturing issues,” said CytRx President and CEO Steven A. Kriegsman. “In addition to obtaining valuable information related to Kaposi’s sarcoma, this trial represents another opportunity to validate the value and viability of our linker technology platform.” The company expects to announce Phase-2 study results in the second quarter of 2015.
Kaposi’s sarcoma is an orphan indication, meaning that only a small portion of the population has been diagnosed with the disease (fewer than 200,000 individuals in the country), and in turn, little research and drug development is being conducted to treat and cure it. The FDA’s Orphan Drug Act may grant orphan drug designation to a drug such as aldoxorubicin that treats a rare disease like Kaposi’s sarcoma, offering market exclusivity for seven years, fast-track status in some cases, tax credits, and grant monies to accelerate research
INNO-206 is an anthracycline in early clinical trials at CytRx Oncology for the treatment of breast cancer, HIV-related Kaposi’s sarcoma, glioblastoma multiforme, stomach cancer and pancreatic cancer. In 2014, a pivotal global phase 3 clinical trial was initiated as second-line treatment in patients with metastatic, locally advanced or unresectable soft tissue sarcomas. The drug candidate was originally developed at Bristol-Myers Squibb, and was subsequently licensed to KTB Tumorforschungs. In August 2006, Innovive Pharmaceuticals (acquired by CytRx in 2008) licensed the patent rights from KTB for the worldwide development and commercialization of the drug candidate. No recent development has been reported for research that had been ongoing for the treatment of small cell lung cancer (SCLC).
INNO-206 is a doxorubicin prodrug. Specifically, it is the 6-maleimidocaproyl hydrazone of doxorubicin. After administration, the drug candidate rapidly binds endogenous circulating albumin through the acid sensitive EMCH linker. Circulating albumin preferentially accumulates in tumors, bypassing uptake by other non-specific sites including the heart, bone marrow and the gastrointestinal tract. Once inside the acidic environment of the tumor cell, the EMCH linker is cleaved and free doxorubicin is released at the tumor site. Like other anthracyclines, doxorubicin inhibits DNA and RNA synthesis by intercalating between base pairs of the DNA/RNA strand, thus preventing the replication of rapidly-growing cancer cells. It also creates iron-mediated free oxygen radicals that damage the DNA and cell membranes. In 2011, orphan drug designation was assigned in the U.S. for the treatment of pancreatic cancer and for the treatment of soft tissue sarcoma.
CytRx Corporation (NASDAQ:CYTR) has announced it has initiated a pivotal global Phase 3 clinical trial to evaluate the efficacy and safety of aldoxorubicin as a second-line treatment for patients with soft tissue sarcoma (STS) under a Special Protocol Assessment with the FDA. Aldoxorubicin combines the chemotherapeutic agent doxorubicin with a novel linker-molecule that binds specifically to albumin in the blood to allow for delivery of higher amounts of doxorubicin (3.5 to 4 times) without several of the major treatment-limiting toxicities seen with administration of doxorubicin alone.
According to a news from Medicalnewstoday.com; CytRx holds the exclusive worldwide rights to INNO-206. The Company has previously announced plans to initiate Phase 2 proof-of-concept clinical trials in patients with pancreatic cancer, gastric cancer and soft tissue sarcomas, upon the completion of optimizing the formulation of INNO-206. Based on the multiple myeloma interim results, the Company is exploring the possibility of rapidly including multiple myeloma in its INNO-206 clinical development plans.
According to CytRx's website, In preclinical models, INNO-206 was superior to doxorubicin with regard to ability to increase dosing, antitumor efficacy and safety. A Phase I study of INNO-206 that demonstrated safety and objective clinical responses in a variety of tumor types was completed in the beginning of 2006 and presented at the March 2006 Krebskongress meeting in Berlin. In this study, doses were administered at up to 4 times the standard dosing of doxorubicin without an increase in observed side effects over historically seen levels. Objective clinical responses were seen in patients with sarcoma, breast, and lung cancers.
INNO-206 - Mechanism of action:
According to CytRx's website, the proposed mechanism of action is as the follow steps: (1) after administration, INNO-206 rapidly binds endogenous circulating albumin through the EMCH linker. (2) circulating albumin preferentially accumulates in tumors, bypassing uptake by other non-specific sites including heart, bone marrow and gastrointestinal tract; (3) once albumin-bound INNO-206 reaches the tumor, the acidic environment of the tumor causes cleavage of the acid sensitive linker; (4) free doxorubicin is released at the site of the tumor.
INNO-206 - status of clinical trials:
CytRx has announced that, in December 2011, CytRx initiated its international Phase 2b clinical trial to evaluate the preliminary efficacy and safety of INNO-206 as a first-line therapy in patients with soft tissue sarcoma who are ineligible for surgery. The Phase 2b clinical trial will provide the first direct clinical trial comparison of INNO-206 with native doxorubicin, which is dose-limited due to toxicity, as a first-line therapy. (source:http://cytrx.com/inno_206, accessed date: 02/01/2012).
Results of Phase I study:
In a phase I study a starting dose of 20 mg/m2 doxorubicin equivalents was chosen and 41 patients with advanced cancer disease were treated at dose levels of 20–340 mg/m2 doxorubicin equivalents . Treatment with INNO-206 was well tolerated up to 200 mg/m2 without manifestation of drug-related side effects which is a ~3-fold increase over the standard dose for doxorubicin (60 mg/kg). Myelosuppression and mucositis were the predominant adverse effects at dose levels of 260 mg/m2 and became dose-limiting at 340 mg/m2. 30 of 41 patients were assessable for analysis of response. Partial responses were observed in 3 patients (10%, small cell lung cancer, liposacoma and breast carcinoma). 15 patients (50%) showed a stable disease at different dose levels and 12 patients (40%) had evidence of tumor progression. (source: Invest New Drugs (2010) 28:14–19)
References
|
1: Kratz F, Azab S, Zeisig R, Fichtner I, Warnecke A. Evaluation of combination therapy schedules of doxorubicin and an acid-sensitive albumin-binding prodrug of doxorubicin in the MIA PaCa-2 pancreatic xenograft model. Int J Pharm. 2013 Jan 30;441(1-2):499-506. doi: 10.1016/j.ijpharm.2012.11.003. Epub 2012 Nov 10. PubMed PMID: 23149257.
2: Walker L, Perkins E, Kratz F, Raucher D. Cell penetrating peptides fused to a thermally targeted biopolymer drug carrier improve the delivery and antitumor efficacy of an acid-sensitive doxorubicin derivative. Int J Pharm. 2012 Oct 15;436(1-2):825-32. doi: 10.1016/j.ijpharm.2012.07.043. Epub 2012 Jul 28. PubMed PMID: 22850291; PubMed Central PMCID: PMC3465682.
3: Kratz F, Warnecke A. Finding the optimal balance: challenges of improving conventional cancer chemotherapy using suitable combinations with nano-sized drug delivery systems. J Control Release. 2012 Dec 10;164(2):221-35. doi: 10.1016/j.jconrel.2012.05.045. Epub 2012 Jun 13. PubMed PMID: 22705248.
4: Sanchez E, Li M, Wang C, Nichols CM, Li J, Chen H, Berenson JR. Anti-myeloma effects of the novel anthracycline derivative INNO-206. Clin Cancer Res. 2012 Jul 15;18(14):3856-67. doi: 10.1158/1078-0432.CCR-11-3130. Epub 2012 May 22. PubMed PMID: 22619306.
5: Kratz F, Elsadek B. Clinical impact of serum proteins on drug delivery. J Control Release. 2012 Jul 20;161(2):429-45. doi: 10.1016/j.jconrel.2011.11.028. Epub 2011 Dec 1. Review. PubMed PMID: 22155554.
6: Elsadek B, Kratz F. Impact of albumin on drug delivery--new applications on the horizon. J Control Release. 2012 Jan 10;157(1):4-28. doi: 10.1016/j.jconrel.2011.09.069. Epub 2011 Sep 16. Review. PubMed PMID: 21959118.
7: Kratz F, Fichtner I, Graeser R. Combination therapy with the albumin-binding prodrug of doxorubicin (INNO-206) and doxorubicin achieves complete remissions and improves tolerability in an ovarian A2780 xenograft model. Invest New Drugs. 2012 Aug;30(4):1743-9. doi: 10.1007/s10637-011-9686-5. Epub 2011 May 18. PubMed PMID: 21590366.
8: Boga C, Fiume L, Baglioni M, Bertucci C, Farina C, Kratz F, Manerba M, Naldi M, Di Stefano G. Characterisation of the conjugate of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin with lactosaminated human albumin by 13C NMR spectroscopy. Eur J Pharm Sci. 2009 Oct 8;38(3):262-9. doi: 10.1016/j.ejps.2009.08.001. Epub 2009 Aug 18. PubMed PMID: 19695327.
9: Graeser R, Esser N, Unger H, Fichtner I, Zhu A, Unger C, Kratz F. INNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma model. Invest New Drugs. 2010 Feb;28(1):14-9. doi: 10.1007/s10637-008-9208-2. Epub 2009 Jan 8. PubMed PMID: 19148580.
10: Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008 Dec 18;132(3):171-83. doi: 10.1016/j.jconrel.2008.05.010. Epub 2008 May 17. Review. PubMed PMID: 18582981.
11: Unger C, Häring B, Medinger M, Drevs J, Steinbild S, Kratz F, Mross K. Phase I and pharmacokinetic study of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin. Clin Cancer Res. 2007 Aug 15;13(16):4858-66. PubMed PMID: 17699865.
12: Lebrecht D, Walker UA. Role of mtDNA lesions in anthracycline cardiotoxicity. Cardiovasc Toxicol. 2007;7(2):108-13. Review. PubMed PMID: 17652814.
13: Kratz F. DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin Investig Drugs. 2007 Jun;16(6):855-66. Review. PubMed PMID: 17501697.
14: Kratz F, Ehling G, Kauffmann HM, Unger C. Acute and repeat-dose toxicity studies of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (DOXO-EMCH), an albumin-binding prodrug of the anticancer agent doxorubicin. Hum Exp Toxicol. 2007 Jan;26(1):19-35. PubMed PMID: 17334177.
15: Lebrecht D, Geist A, Ketelsen UP, Haberstroh J, Setzer B, Kratz F, Walker UA. The 6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH) is superior to free doxorubicin with respect to cardiotoxicity and mitochondrial damage. Int J Cancer. 2007 Feb 15;120(4):927-34. PubMed PMID: 17131338.
16: Di Stefano G, Lanza M, Kratz F, Merina L, Fiume L. A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: synthesis, characterization and preliminary biological properties of the conjugate. Eur J Pharm Sci. 2004 Dec;23(4-5):393-7. PubMed PMID: 15567293.
| EP0169111A1 * | Jun 18, 1985 | Jan 22, 1986 | Sanofi | Cytotoxic conjugates useful in therapy, and process for obtaining them |
| EP0269188A2 * | Jun 18, 1985 | Jun 1, 1988 | Elf Sanofi | Cytotoxic conjugates useful in therapy, and process for obtaining them |
| EP0306943A2 * | Sep 8, 1988 | Mar 15, 1989 | Neorx Corporation | Immunconjugates joined by thioether bonds having reduced toxicity and improved selectivity |
| EP0328147A2 * | Feb 10, 1989 | Aug 16, 1989 | Bristol-Myers Squibb Company | Anthracycline immunoconjugates having a novel linker and methods for their production |
| EP0398305A2 * | May 16, 1990 | Nov 22, 1990 | Bristol-Myers Squibb Company | Anthracycline conjugates having a novel linker and methods for their production |
| EP0457250A2 * | May 13, 1991 | Nov 21, 1991 | Bristol-Myers Squibb Company | Novel bifunctional linking compounds, conjugates and methods for their production |
Subscribe to:
Posts (Atom)