Tracks information on drugs on worldwide basis by Dr Anthony Melvin Crasto, helping millions with websites, 9 million hits on google, 2.5 lakh connections worldwide, P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Monday, 24 February 2014
Sunday, 23 February 2014
RAMELTEON, TAK 375 ..Melatonin MT1/MT2 receptor agonist
Feb232014

RAMELTEON
ACN-S001714, ZINC00007031
- HSDB 7787
- Ramelteon
- Rozerem
- TAK-375
- UNII-901AS54I69
Molecular Formula: C16H21NO2 Molecular Weight: 259.34344
| CAS number | 196597-26-9 |
|---|
| (S)-N-[2-(1,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)ethyl]propionamide |
(5)-N-[2-(l,6,7,8-tetrahydro-2H-indeno-[5,4-ό]furan-8- yl)ethyl]propionamide
| United States | US 6034239 | 1999-07-22 | expiry 2019-07-22 |
EP885210B1 , EP1792899A1 and J. Med Chem. 2002, 45, 4222-4239
NMR
- dmd.aspetjournals.org/content/suppl/.../Supplemental_Information.pptxMay 17, 2010 - Ramelteon NMR Assignments. COSY: Black Arrows. HMBC: Red Arrows. Figure S-1b. 1H NMR Spectrum of Ramelteon. Figure S-1c.
Ramelteon is the first in a new class of sleep agents that selectively binds to the melatonin receptors in the suprachiasmatic nucleus (SCN). It is used for insomnia, particularly delayed sleep onset. Ramelteon has not been shown to produce dependence and has shown no potential for abuse.
Ramelteon, marketed as Rozerem by Takeda Pharmaceuticals North America, is the first in a new class of sleep agents that selectively binds to the MT1 and MT2 receptors in the suprachiasmatic nucleus (SCN), instead of binding to GABA A receptors, such as with drugs like zolpidem,eszopiclone, and zaleplon. Ramelteon is approved by the U.S. Food and Drug Administration (FDA) for long-term use.
Ramelteon does not show any appreciable binding to GABAA receptors, which are associated with anxiolytic, myorelaxant, and amnesic effects.
Rozerem (ramelteon), FDA Approved 07.04.05, can be used for insomnia, particularly delayed sleep onset. Ramelteon has not been shown to produce dependence and has shown no potential for abuse, and the withdrawal and rebound insomnia that is typical with GABA modulators is not present in ramelteon. Some clinicians also use ramelteon for the treatment of Delayed sleep phase syndrome.
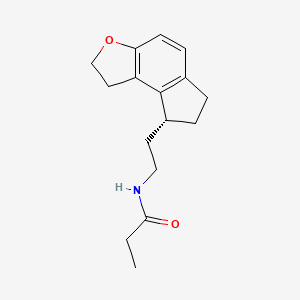 Ramelteon
RamelteonMechanism of action
Ramelteon is a melatonin receptor agonist with both high affinity for melatonin MT1 and MT2 receptors and selectivity over the MT3 receptor. Ramelteon demonstrates full agonist activity in vitro in cells expressing human MT1 or MT2 receptors, and high selectivity for human MT1 and MT2receptors compared to the MT3 receptor.[1]
The activity of ramelteon at the MT1 and MT2 receptors is believed to contribute to its sleep-promoting properties, as these receptors, acted upon by endogenous melatonin, are thought to be involved in the maintenance of the circadian rhythm underlying the normal sleep-wake cycle. Ramelteon has no appreciable affinity for the GABA receptor complex or for receptors that bind neuropeptides, cytokines, serotonin, dopamine, noradrenaline,acetylcholine, and opiates. Ramelteon also does not interfere with the activity of a number of selected enzymes in a standard panel.
The significance of ramelteon’s lack of affinity for the MT3 receptor is not clear, despite the manufacturer’s emphasis of this fact in commercial advertisements. The MT3 receptor appears almost exclusively in the gut and might not have any relationship to sleep or wakefulness.

The major metabolite of ramelteon, M-II, is active and has approximately one tenth and one fifth the binding affinity of the parent molecule for the human MT1 and MT2 receptors, respectively, and is 17 – 25-fold less potent than ramelteon in in vitro functional assays. Although the potency of M-II at MT1 and MT2 receptors is lower than the parent drug, M-II circulates at higher concentrations than the parent producing 20 – 100 fold greater mean systemic exposure when compared to ramelteon. M-II has weak affinity for the serotonin 5-HT2B receptor, but no appreciable affinity for other receptors or enzymes. Similar to ramelteon, M-II does not interfere with the activity of a number of endogenous enzymes.
All other known metabolites of ramelteon are inactive.
No published studies have indicated whether ramelteon, in humans, is more or less safe or effective than the hormone melatonin which it mimics; melatonin is much less expensive and is widely available over-the-counter in the US and Canada. The biological action of melatonin is similar to that of ramelteon. Ramelteon has been directly compared to melatonin in cats, and Ramelteon had a significant (3x) longer effect and had a more profound effect on the EEG of the sleeping cats.[2]
Introduction
ROZEREM (ramelteon) is an orally active hypnotic, chemically designated as (S)-N-[2- (l,6,7,8-tetrahydro-2H-indeno-[5,4-b]furan-8-yl)ethyl]propionamide, and contains one chiral center. The compound is produced as the (S)-enantiomer, with an empirical formula of C16H21N02, molecular weight of 259.34, and the following chemical structure (I):

(I) -Ramelteon
Ramelteon is used to help patients who have sleep-onset insomnia (difficulty falling asleep) to fall asleep more quickly. It is the first in a new class of sleep agents that selectively binds to the MT] and MT2 receptors in the suprachiasmatic nucleus (SCN), in a class of medications called melatonin receptor agonists with both high affinity for melatonin MT! and MT2 receptors and selectivity over the MT3 receptor. It works similarly to melatonin, a natural substance in the brain that is needed for sleep.
Ramelteon was first disclosed in European patent EP 885210, which also disclosed a process for synthesizing ramelteon, as shown in scheme 1 : Scheme 1

Ramelteon
The processes of the prior art suffer from many disadvantages, some of which result from the fact that they involve several steps.
For instance, in US patent US 6034239, which is related to EP 885210, there is disclosed a process for preparing an intermediate compound of Formula (IV), which involves conversion of diethylcyano methyl phosphonate in the presence of 60% sodium hydride. Disadvantages of this particular reaction include the need for the highly flammable and corrosive base sodium hydride, the use of toxic triethyl phosphate for the formation of diethylcyano methyl phosphonate (which also has a high boiling point), and low yield of 60%. Such disadvantages mean that the disclosed process is difficult to implement industrially or economically. A further problem associated with prior art preparation techniques is the formation of dimeric impurities at the nitrile reduction stage (i.e. where the intermediate of Formula (IV) is reduced). For instance, US 6034239 discloses reduction of (l,2,6,7-Tetrahydro-8H-indeno-[5,4- b]furan-8-ylidene)-acetonitrile of formula (IV) by means of H2 over Raney nickel in in a solvent medium of ethanol NH3 to provide compound of formula (IIA). The reaction is carried out by applying 5 kg of hydrogen pressure, which results in the formation of the byproduct and impurity Dimer A, which in turn affects the yield and purity of the product of formula (IIA).

Dimer A
Similarly, (l,2,6,7-Tetrahydro-8H-indeno-[5,4-b]furan-8-ylidene)-acetonitrile of formula (IV) may be reduced by means of H2 over Raney cobalt in a solvent medium of ethanol/ NH3 to afford compound of formula (IIB). The reaction, which is carried out by applying hydrogen pressure, is not selective, and results in the formation of the by-product and impurity Dimer B, which in turn affects the yield and purity of product of formula (IIB).

Dimer B
Repeated purifications are required to remove impurities such as Dimer A and B to obtain ramelteon having the desired purity, which results in the poor yield of ramelteon.
Several other approaches are also described in the literature to make ramelteon and related compounds in WO2006030739, WO208062468, WO2008106179, US 2010152468, WO2009106966 and WO2010/055481. However, all processes of the prior art for the preparation of ramelteon are cumbersome; the processes employ a plurality of reagents and involve multiple steps, which make the overall processes uneconomical. Therefore there is a need for a more economical, efficient and industrially suitable method of making ramelteon, whereby address the problems associated with prior art, some of which are discussed above.
Ramelteon is the active ingredient in trademarked ROZEREM®, and is approved by the United States Food and Drug Administration for the treatment of insomnia characterized by difficulty with sleep onset.
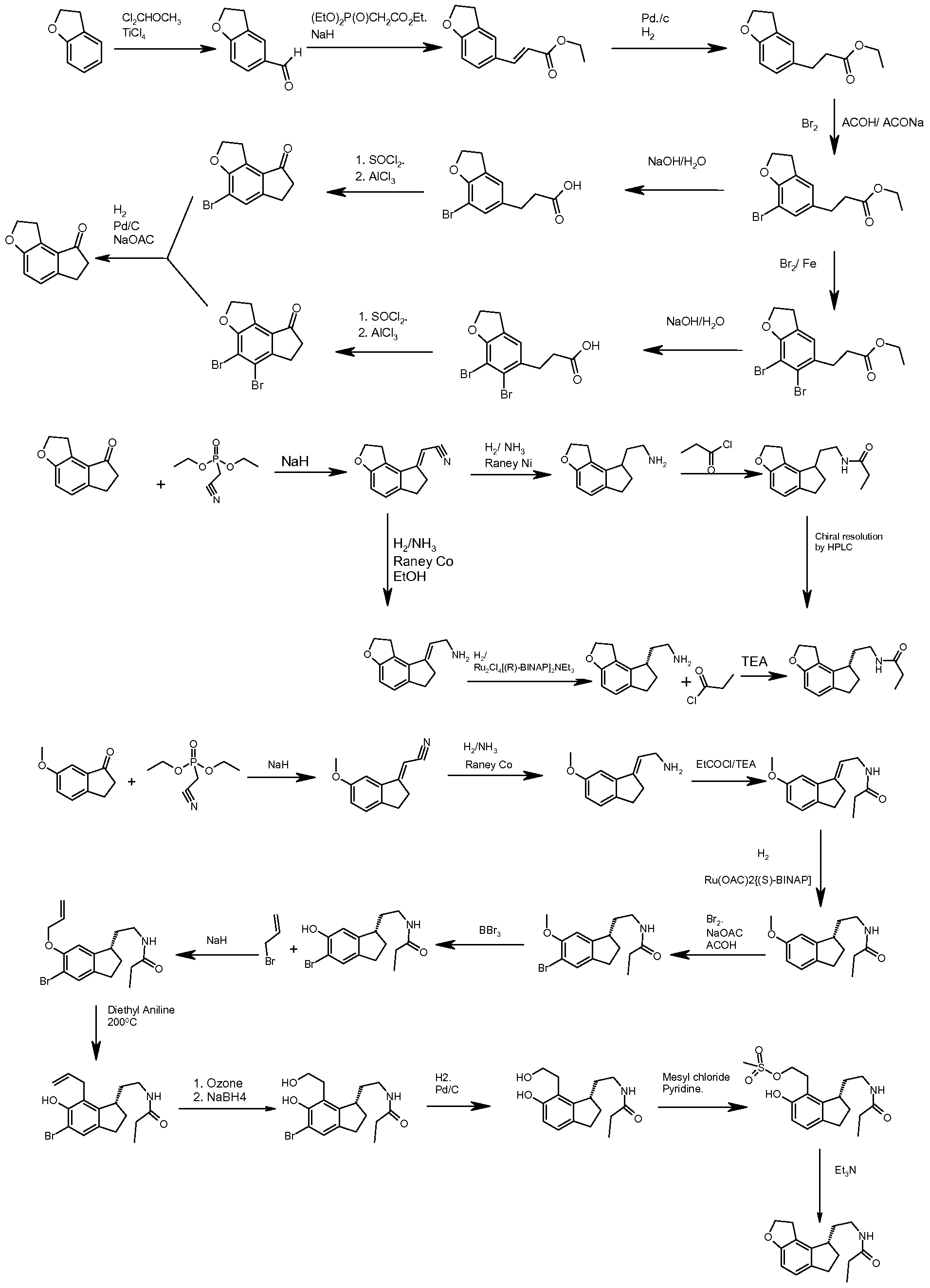
Different processes for preparing (S)-N-[2-(l,6,7,8-tetrahydro-2H-indeno-[5,4- b]furan-8-yl)ethyl]propionamide, i.e. ramelteon, are disclosed in US 6034239, JP 11080106, JP 11140073 and WO 2006/030739. U.S. Patent No. 6034239 describes the following processes for the preparation of ramelteon:
Japan Patent Publication No. 11080106 reports the following process for the preparation of ramelteon:
Ru(OCOCH3)[(R)-BI NAP] IOOatm H2/50 temp


BF3 DEE Complex

Japan Patent Publication no. 11140073 reports the following process for the preparation of an intermediate of ramelteon:

PCT Publication No. WO/2006/030739 reports the following process for the preparation of ramelteon:
POCI3/DMF (EtO)2P(O)CH2CO2Et Toluene NaH/Toluene







Purification in Ethanol water 95 7%
United States Patent No. 6,034,239 discloses the formation of chiral intermediates (S)-(- )-N-[2-(l,6,7,8,-tetrahydro-2H-indeno[5,4-b]furan-8-yl)ethylamine (sometimes referred to as compound S-2 or intermediate compound S-2) by the catalytic asymmetric hydrogenation of 2- (l,2,6,7,-tetrahydro-8H-indeno[5,4-b]furan-8-ylidene)ethylamine (compound 3 in the reaction scheme shown below) in the presence of a catalytic amount of BINAP-ruthenium complex in approximately 89% e.e. (enantiomeric excess). Following the catalytic reaction, the product is purified by preparing acid salts and acylated with propionyl chloride (compound 4 in the reaction scheme shown below) to obtain ramelteon (compound 1 in the reaction scheme shown below) in its pure (S) isomer form.

|S)*2
An alternate process for preparing ramelteon is disclosed in the Journal of Medicinal Chemistry, Vol. 45, pp. 4222-4239 (2002), wherein the exo double bond of intermediates (A) shown below was asymmetrically reduced using (S)-2, 2′-bis-(diphenylphosphino)-l, 1 ‘- binaphthyl (binap)-Ru complex as the catalyst to obtain the enantiomerically pure compound (B). Compound (B) is subsequently converted to ramelteon (1) through the intermediate steps of Claisen condensation, ozonolysis and cyclization.

m Both of the above processes uses expensive catalyst and give poor enantioselectivity. Additionally, these processes are expensive due to the need to perform multiple purifications steps in order to achieve an enantioselectivity of at least about 99% or greater of the desired isomer.
PCT Patent Publication No. WO 2008/062468 A2 discloses the following process for the preparation of ramelteon:

RAMELTEON
WO 2008/062468 teaches that separation of the enantiomers of intermediate (2) may be accomplished by: i) optical resolution of the racemic amine intermediate (2) by preparing acid salts with chirally pure acids; or ii) chromatographic techniques using chiral and/or achiral stationary phases for batch process, super critical or sub critical chromatography and/or continuous process chromatography. Although WO 2008/062468 mentions the possible use of optical resolution with chirally pure acids, there is no further teaching, discussion or disclosure of this method. WO 2008/062468 does, however, provide detailed descriptions of chromatographic methods for separating the isomers of intermediate compound (2). The disclosed chromatographic process suffers the following disadvantages:
• Preparative chromatography is time consuming & expensive;
• Highly sophisticated instrumentation required; • Not commercially feasible.
PCT Patent Publication No. WO 2008/106179 discloses a process for the preparation of ramelteon that involves the following reaction steps:

wherein X= O-alkyl or NH2 and chiral reduction of the compound of formula IV in the presence of Ru-BINAP complex under hydrogen atmosphere in an organic solvent.

IV V
The process disclosed in WO 2008/106179 is similar to the process disclosed in United States Patent No. 6,034,239 and the Journal of Medicinal Chemistry, Vol. 45 in that a Ru-BINAP complex is employed.
Resolution of racemic mixtures via reaction with optically active acids and the subsequent crystallization of the resulting salts is preferably employed when the chiral carbon of the racemic compound is an alpha carbon {i.e., one carbon removed) to the functional group forming the acid addition salt. As the distance between the chiral carbon of the racemic compound to the functional group of the racemic compound increases to beta (i.e., two carbon removed) & gamma (i.e., three carbon removed), the resolution of the diastereomeric salt becomes more difficult and not very useful.
Ramelteon has a chiral center at the gamma carbon, which makes the separation of the isomer with an optically active acid quite a daunting task. Similarly, N-[2-(l, 6, 7, 8,- tetrahydro-2H-indeno [5, 4-b]furan-8-yl)]ethylamine (compound T), an intermediate useful in the production of ramelteon has a chiral center at the gamma carbon which would lead a skilled artisan to believe that optical resolution with an optically active acid could prove difficult.
Synthesis

Chilman-Blair, K.; Castañer, J.; Silvestre, J.S.; Bayés, M. (2003). “TAK-375″. Drugs of the Future 28 (10): 950. doi:10.1358/dof.2003.028.10.763214.
………………..
SYNTHESIS
Scheme 1 :

XIV (S)-XII

……………………………………..
SYNTHESIS
Scheme 2

0-30°C
Metal salt Propionyl halide/
Propionc anhydride

Ramelteon (I)
Synthesis of ramelteon
Preparation 1
N-[2-(l,6,7,8-Tetrahydro-2H-indeno[5,4-b]furan-8-yl)-ethyl]-propionamide (2.0 gm) was dissolved in 50.0 ml (n-Hexane:IPA:DEA) (as used herein, “IPA” stands for isopropyl alcohol, and “DEA” stands for diethylamine)
and optically resolved by high performance column chromatography on CHTRAL PACK IA-3 using Mobile phase : n-Hexane:IPA:DEA Flow rate: 1.0ml/min UV:285 nm; at a column temperature of 25°C;sample concentartion: lmg/ml and, eluted with mobile phase. Both the enantiomers were collected separately and after evaporation of solvent under vacuum, enantiomerically pure ramelteon (I) was obtained. Preparation 2- using Supercritical Fluid Chromatography (SFC)
N-[2-(l56,7,8-Tetrahydro-2H-indeno[5,4-b]furan-8-yl)-ethyl]-propionamide (2.0 gm) was dissolved in 50.0 ml (n-Hexane:Ethanol:DEA) and optically resolved by Supercritical Fluid Chromatography (SFC) on CHIRAL PACK AD-H using a mobile phase : C02/(Methanol/ Diethylamine[DEA]) and eluted with mobile phase. Both the enantiomers were collected separately and after evaporation of solvent under vacuum, enantiomerically pure S- ramelteon of Formula (I) and R-ramelteon were obtained with isomeric purity>99%.
………………………………
SYNTHESIS
synthesis of ramelteon that comprises the step of separating N-[2-(l, 6, 7, 8-tetrahydro-2H-indeno [5, 4-b] furan-8- yl)]ethylamine (compound 2) into its isomers using an optically active acid to achieve high enantioselectivity of the desired isomer. This embodiment may further include the step of acylating the substantially pure enantiomer, (S)-N-[2-(l, 6, 7, 8-tetrahydro-2H-indeno [5, 4-b] furan-8-yl)]ethylamine (compound (S)-2) using a suitable acylating agent, such as propionyl chloride) to provide (S)-7V-[2-(l,6,7,8-tetrahydro-2H-indeno[5,4-b]furan-8-yl]ethyl]propionamide (ramelteon or compound 1) substantially free of the (R)-isomer.
One embodiment of the present invention for the preparation of ramelteon is shown below in Scheme 1.

Example 1
Preparation of (S)-N-2-(l, 6, 7, 8-tetrahydro-2H-indeno [5, 4-b] furan-8-yl) ethylamine (Compound (S)-2)
A solution of N-[2-(l, 6, 7, 8-tetrahydro-2H-indeno [5, 4-b] furan-8-yl)ethylamine (45 g; 0.22 mol) in methanol (225 ml) is added to a solution of S-(+)-2-(4-isobutylphenyl)propionic acid (41 g; 0.20 mol) in methanol (205 ml) at 25-300C. The reaction mixture is concentrated to dryness under reduced pressure. The crude salt precipitated is recrystallized in methanol to give a diastereomeric salt of (S)-N-2-(l, 6, 7, 8-tetrahydro-2H-indeno [5, 4-b] furan-8-yl) ethylamine with (S)-(+)-2-(4-isobutylphenyl) propionic acid having a chiral purity of greater than 90% enantioselectivity. The product obtained is recrystallized from methanol to give the pure salt having chiral purity of 99% or greater enantioselectivity.
The purified salt is suspended in water and the pH of the suspension is adjusted to 11-12 using aqueous sodium hydroxide. The reaction mixture is extracted with dichloromethane, washed with water and evaporated to give the pure (S)-N-[2-(l, 6, 7, 8-tetrahydro-2H-indeno [5, 4-b] furan-8-yl)]ethylamine (compound (S)-2), substantially free from its (R) isomer.
Example 2
Preparation of (S)-N-[2-(l, 6, 7, 8-tetrahydro-2H-indeno [5, 4-b] furan-8-yl)ethyl] propionamide (ramelteon)
Triethyl amine (15.15 g, 0.15 mol) and propionyl chloride (13.66 g, 0.15 mol) were added to a solution of S-[2-(l, 6, 7, 8-tetrahydro-2H-indeno [5,4-b]furan-8-yl)]ethylamine (25 g, 0.12 mol) (compound (S)-2) (prepared in Example 1) in dichloromethane and stirred at room temperature for 2 hours. 75 mL water was added to the reaction mixture, and the layers were separated. The dichloromethane layer was concentrated under reduced pressure and purified from a mixture of acetone and hexane to give (S)-N-[2-(l, 6, 7, 8-tetrahydro-2H-indeno [5, 4-b] furan- 8-yl) ethyl] propionamide (compound 1) having a chiral purity of 99% or greater enantioselectivity.
…………
INTERMEDIATES
The intermediate compound of formula Vl, 6,7-dihydro-1 H-indeno[5,4-b]furan-8(2H)-one, can then be subjected to further synthesis steps to yield ramelteon by synthesis route known to or readily devisable by a person skilled in the art, suitably involving the introduction of the side chain having chirality and amide function. The documents mentioned infra are incorporated herein by way of reference. For example, the following synthesis route may be applied:

1 ) NaOH
Vl 2) H2, Ru-BI NAP
3) HCI
4) H2, Pd/C

Experimental Procedures
Example 1 :
Preparation of 4-(2-chloroethyl)-2,3-dihydrobenzofuran (II)

intermediate FTIR spectra of MeCN (140 ml) was recorded as reference. MeCN was cooled to -200C, oxalyl chloride (16.5 ml) was added at once and waited until temperature re-stabilized at – 200C. DMF (16.6 ml) was then added drop-wise (temperature between -18°C and -22°C, 0.5 ml/min). Reaction was stirred until no oxalyl chloride was visible and DMF level was stable by FTIR. Vilsmeier reagent is thereby formed in situ according to the following reaction:

Product I was then added portion wise (temperature between -18°C and -210C, about 30 min). Formation of intermediate was immediately observed by FTIR. Reaction was stirred for one hour. Et3N was then added drop-wise (temperature between -18°C and -22°C, 50 ml/h). At the end of addition, reaction was stirred 15 min at -200C and temperature was slowly raised to 500C (within about 15 min). Disappearance of intermediate and formation of DMF and product Il was monitored by FTIR. When reaction looked completed by FTIR (about 2h at 50°C), the reaction was cooled down to 200C and quenched with water (45 ml). Solution was transferred to a round bottom flask and MeCN was removed under reduced pressure. Solution was then diluted with MTBE (100 ml) and water (50 ml). Phases were separated and aqueous phase was re-extracted twice with MTBE (50 ml). Combined organic phases were washed twice with 10% H3Pθ4/10% NaCI solution and stored at 4°C until next step.
List of FTIR bands used to follow the reaction (using 2nd derivative and solvent subtraction): Oxalyl chloride (reactant): Height to two point baseline, peak from 1800 cm“1 to 1770 cm“1, baseline 1800 cm“1 to 1770 cm“1.
Intermediate: Height to single point baseline, peak from 1722 cm“1 to 1712 cm“1, baseline
1722 cm“1.
Compound Il (product): Area to two point baseline, peak from 993 cm“1 to 981 cm“1, baseline 993 cm“1 to 981 cm“1.
DMF: Height to single point baseline, peak from 1694 cm“1 to 1680 cm“1, baseline 1694 cm“1.
Example 2:
Preparation of 4-vinyl-2,3-dihydrobenzofuran (III)

M FTIR spectra of MTBE was recorded prior to the reaction as reference. To the solution of 4- (2-chloroethyl)-2,3-dihydrobenzofuran (II) in MTBE (150 ml) obtained at the previous step, was added, water (38 ml), Kl (1.37 g), Bu4NOH 40% (19 ml) and NaOH 50% solution (66 ml). Reaction was vigorously stirred and heated at 500C until reaction looked completed by FTIR (4 to 5 h). Warm reaction mixture was then transferred into an extraction funnel to give three phases. Water phase (bottom) was removed and did not contain product. Medium phase (colored black) was diluted with water (120 ml) and was extracted three times with MTBE. Combined organic phases were washed twice with water, once with 0.5M NaHSO3/10% NaCI solution and once with 1 N NaOH/10% NaCI solution. MTBE solution was dried using MgSO4, filtered, concentrated and used immediately for next step.List of FTIR bands used to follow the reaction (using 2nd derivative and solvent subtraction) Compound Il (reactant): Area to zero, peak from 1440 cm“1 to 1437 cm“1 Compound III (product): Area to zero, peak from 1417 cm“1 to 1412 cm“1. Compound III (product): Area to zero, peak from 1565 cm“1 to 1562 cm“1.
Example 3:
Preparation of 1-(2,3-dihydrobenzofuran-4-yl)ethanone (V)

4-vinyl-2,3-dihydrobenzofuran (I I I ) (2.4 g) was dissolved in toluene (2 ml) and were successively added (ITC) (51 mg), PdCI2(30 mg) and H2O2 30% (2 ml). Reaction was vigorously stirred at 55°C until reaction looked completed by FTIR. (for around 24 h). Reaction was cooled down to room temperature, diluted with EtOAc (50 ml) and water (50 ml). Phases were separated and organic phase was washed with 0.5M NaHSO3/10% NaCI solution and twice with 1 M NaHCO3, dried over MgSO4 and concentrated. Purification by flash chromatography gave 1-(2,3-dihydrobenzofuran-4-yl)ethanone (V). 1H NMR δ (CDCI3) 7.35 (dd, 1 H, J = 0.8 Hz, J = 7.8 Hz), 7.19 (t, 1 H, J = 7.9 Hz), 6.95 (d, 1 H, J = 8.0 Hz), 4.57 (t, 2H, J = 8.8 Hz), 3.52 (t, 2H, J = 8.8 Hz), 2.57 (s, 3H). 13C NMR δ (CDCI3) 198.8, 161.0, 133.8, 128.2, 127.9, 121.4, 1 13.4, 71.6, 31.0, 27.6.
List of FTIR bands used to follow the reaction (using 2nd derivative and solvent subtraction) Compound III (reactant): Area to single point baseline, peak from 925 cm“1 to 915 cm“1, baseline 915 cm“1.
Compound V (product): Area to zero, peak from 1730 cm“1 to 1724 cm“1.
Example 4:
Preparation of 6,7-dihydro-1 H-indeno[5,4-b]furan-8(2H)-one (Vl)

V Vl1 -(2,3-dihydrobenzofuran-4-yl)ethanone (V) (1 g, 6.2 mmol) was dissolved in dioxane (9 ml). TADCA (dicyclohexylammonium 2,2,2-trifluoroacetate) (1 .82 g, 1 eq) and paraformaldehyde (0.61 1 g, 1.1 eq) were added. The reaction was heated at 1000C for 2 h. A second portion of TADCA (0.91 g, 0.5 eq) and paraformaldehyde (0.333 g, 0.6 eq) were added and the reaction was heated at 1000C for 2 h. Reaction was partitioned between water (20 ml) and pentane (30 ml). Aqueous phase was re-extracted 4 times with pentane (10 ml). Combined pentane phases were washed with water and brine, dried over MgSO4. Solution was diluted to 100 ml with pentane. This solution was added dropwise to a pre-heated solution of sulfuric acid at 67°C (10 ml) under nitrogen stream. At the end of addition, the reaction was stirred for 30 min. Reaction was cooled down to room temperature and poured on iced water (50 ml). Solution was extracted 5 times with MTBE. Combined organic phases were washed with water, NaHCO3 1 M and brine, dried over MgSO4 and concentrated. Purification by flash chromatography furnished pure 6,7-dihydro-1 H-indeno[5,4-b]furan-8(2H)-one (Vl). 1H NMR δ (CDCI3) 7.21 (dd, 1 H, J = 0.9 Hz, J = 9.0 Hz), 7.02 (d, 1 H, J = 8.2 Hz), 4.66 (t, 2H, J = 8.9 Hz), 3.48 (t, 2H, J = 8.9 Hz), 3.08 (dd, 2H, J = 4.9 Hz, J = 6.0 Hz), 2.69 (m, 2H). 13C NMR δ (CDCI3) 207.5, 160.2, 147.1 , 133.6, 125.6, 123.9, 1 15.6, 72.3, 37.1 , 28.4, 25.4.
…………………………………
SYNTHESIS
Improve the synthesis and flow properties of an insomnia drug.
Ramelteon (1), marketed as Rozerem by Takeda Pharmaceuticals, is used to treat insomnia. V. K. Kansal and co-inventors describe several processes that are used to prepare it, all of which require many steps. The inventors offer no comments about the relative merits of the processes, but they state that a new industrial-scale process is needed. Their main claims are to intermediate acid 2 as a racemic mixture and individual enantiomers; one enantiomer is converted to 1 by the route shown in Figure 1.

The inventors use diastereomeric crystallization to resolve the racemic mixture by forming its (S)-1-phenylethylamine salt. The salt of the (R)-isomer of 2 is recovered first; then the salt of (S)-2 is isolated from the solution and acidified to give the free acid, which is purified by using (R)-1-phenylethylamine. Both enantiomers are isolated with >99.0% purity and >99.0% ee.
The (S)-acid is converted to acid chloride 3 and then to amide 4 by reactions with SOCl2and NH3 gas, respectively. The chloride is not isolated; the amide is recovered in 85–90% yield with 95–98% purity. When aq NH4OH is used instead of NH3 gas, the purity of 4 is slightly lower (93–96%). An alternative method for preparing 4 is to treat 2 with Et3N and ClCO2Et, followed by NH3. This method produces 4 in yields of 80–95% and 97–99% purity.
Amide 4 is reduced to amine 5 with NaBH4 and BF3·Et2O. The amine is purified by forming its chloride or oxalate salt in yields as high as 85% and 96–98% purity. The salts are used to prepare 1 by treating them with EtCOCl in the presence of base: NaOH for the chloride salt and Na2CO3 for the oxalate. In both cases, the yield of 1 is >92%, and the purity is as high as 99.9% after recrystallization from EtOH.
The inventors also recrystallized 1 from toluene to produce what they describe as a “nonelectrostatic” crystalline form, designated as form A. They describe the measurement of the electrostatic charge of the crystals in one of the patent’s examples. The measurements show that the average charge density of form A is ≈15 times lower than crystals obtained from EtOAc. Low electrostatic charge improves the flow characteristics of the solid, which is important in preparing drug formulations.
The inventors report the details of preparing rac-2 by a multistep procedure shown in Figure 2.

In most of the reaction steps, the product is isolated in crude form; the inventors do not indicate whether the product is purified before it is used in the next stage. The synthesis of rac-2 begins with the conversion of benzofuran (6) to aldehyde7 by treatment with POCl3 followed by hydrolysis. The crude product is isolated as a liquid in 85–90% yield and 90–92% purity.
In the next step, 7 is condensed with malonic acid (8) in the presence of piperidine and HOAc; acid 9 is isolated in 92–95% yield and 95% purity. Catalytic hydrogenation of 9produces 10 in 95% yield and 94–96% purity. The hydrogenation also can be carried out in the presence of NaOH and HCO2NH4; the yield and purity of 10 are the same, but the reaction takes 6 h instead of 2 h. [The patent does not state why NaOH and HCO2NH4 would be used.—Ed.] Acid 10 is brominated to produce acid 11, isolated in 50–60% yield and 92–95% purity.
The next stage begins with treating 11 with SOCl2 to activate the carboxyl group by forming acid chloride 12. The chloride is not isolated but cyclized under Friedel–Crafts conditions to give tricyclic compound 13, isolated in yields of 85–92% and 90-95% purity. This reaction also produces two impurities, 14 and 15, but the amounts are not reported. Removing the impurities gives 13 in good yield, but the inventors do not describe how this is done. They do report that the impurities can be isolated, and 1H and 13C NMR data are provided for both.
In the next step, the bromine atoms in 13 are replaced by hydrogen to give 16 in 85–90% yield and 96–97%purity. This reaction produces two impurities, 17 and 18; again, the amounts are not reported, but 1H and 13C NMR data are. After MeOH reflux in the presence active carbon, 16 is isolated in 80–85% yield with 99.3–99.8% purity. It is then converted to ester 20 by treating it with a solution of phosphonate 19 that contains suspended NaH. Crude product 20 is isolated in 80–85% yield and 92–95% purity as a mixture of (E)- and (Z)-isomers. The isomer mixture is hydrogenated, and base hydrolysis gives rac-2 in 90–95% isolated yield and 95–98% purity.
The inventors claim that the overall process is suitable for producing ramelteon on an industrial scale in a crystalline form that has improved flow characteristics. (Teva Pharmaceutical Industries [Petah Tiqva, Israel]. US Patent 8,084,630, Dec. 27, 2011;
 RAMELTEON
RAMELTEON
…………………….
SYNTHESIS
CHINESE CHEMICAL LETTERS 22, 2011, 264 SEE SYN OF KEY INTERMEDIATE
1:RAMELTEON
[a]D20 10.0 (c, 0.20, EtOH); mp 76–77 8C;
1H NMR (500 MHz, CDCl3): d1.15 (t, 3H, J = 7.5 Hz), 1.60 (m, 1H), 1.70 (m, 1H), 2.02 (m, 1H), 2.19 (q, 2H, J = 7.5 Hz), 2.32 (m, 1H), 2.76 (m, 1H), 2.85 (m, 1H), 3.11 (m,1H), 3.41 (m, 2H), 3.79 (s, 3H), 5.48 (s, 1H), 6.71 (dd, 1H, J = 2.0 Hz, 8.5 Hz), 6.75 (s, 1H), 7.11 (d, 1H, J = 8.0 Hz).
13C NMR (100 MHz,DMSO–d6): d173.7, 158.7, 148.1, 135.8, 124.9, 112.3, 109.2, 55.5, 42.7, 37.9, 34.8, 32.5, 30.6, 29.8, 9.9. EI-MS: 247 ([M]+); HR-MS 247.1572([M]+
, C15H21NO2; Calcd. 247.1571). The enantiomeric excess of (S)-1 was determined by HPLC as >99.9% [column, CHIRALPAK AS-H
(4.6 mm 250 mm), room temperature; eluent, hexane-2-propanol-trifluoroacetic acid (90:10:0.1); flow rate, 1.0 mL/min; detect, 290 nm; tRof (S)-1, 30.7 min; tR of (R)-1 (enantiomer of (S)-1), 37.1 min].
, C15H21NO2; Calcd. 247.1571). The enantiomeric excess of (S)-1 was determined by HPLC as >99.9% [column, CHIRALPAK AS-H
(4.6 mm 250 mm), room temperature; eluent, hexane-2-propanol-trifluoroacetic acid (90:10:0.1); flow rate, 1.0 mL/min; detect, 290 nm; tRof (S)-1, 30.7 min; tR of (R)-1 (enantiomer of (S)-1), 37.1 min].
…………………….
NMR
- dmd.aspetjournals.org/content/suppl/.../Supplemental_Information.pptxMay 17, 2010 - Ramelteon NMR Assignments. COSY: Black Arrows. HMBC: Red Arrows. Figure S-1b. 1H NMR Spectrum of Ramelteon. Figure S-1c.
References
- Owen RT (April 2006). "Ramelteon: profile of a new sleep-promoting medication". Drugs Today 42 (4): 255–63. doi:10.1358/dot.2006.42.4.970842. PMID 16703122.
- Miyamoto M, Nishikawa H, Doken Y, Hirai K, Uchikawa O, Ohkawa S (November 2004). "The sleep-promoting action of ramelteon (TAK-375) in freely moving cats". Sleep 27 (7): 1319–25.PMID 15586784.
- Zammit G, Erman M, Wang-Weigand S, Sainati S, Zhang J, Roth T (August 2007). "Evaluation of the Efficacy and Safety of Ramelteon in Subjects with Chronic Insomnia". J Clin Sleep Med 3 (5): 495–504. PMC 1978328. PMID 17803013.
- Daniel F. Kipke, MD |title=Evidence That New Hypnotics Cause Cancer |journal=University of California |date=March 2008 |url=http://escholarship.org/uc/item/12r2f32g#page-2
- http://db.wdc-jp.com/cgi-bin/psj/data/cpb/pdf/201108/c08_1062.pdf
- http://sat.ecnu.edu.cn/Uploadnews/20120213113859628.pdf
- https://docs.google.com/viewer?url=http%3A%2F%2Fdmd.aspetjournals.org%2Fcontent%2Fsuppl%2F2010%2F05%2F17%2Fdmd.110.034009.DC1%2FSupplemental_Information.pptx
Full-Text PDF - MDPI.com
- Rozerem Official Website
- Prescribing Information Data Sheet
- RxList.com
- Clinical Pharmacokinetic Monitoring of Midazolam in Critically Ill Patients in relation to midazolam as a drug-drug interaction to Rozerem
| WO2008106179A1 | Feb 26, 2008 | Sep 4, 2008 | Teva Pharma | Intermediates and processes for the synthesis of ramelteon |
| WO2008151170A2 | Jun 2, 2008 | Dec 11, 2008 | Teva Pharma | Process for the synthesis of ramelteon and its intermediates |
| EP0885210B1 | Mar 5, 1997 | Jun 12, 2002 | Takeda Chemical Industries, Ltd. | Tricylic compounds having binding affinity for melatonin receptors, their production and use |
| EP1792899A1 | Sep 12, 2005 | Jun 6, 2007 | Takeda Pharmaceutical Company Limited | Process for production of optically active amine derivatives |
| US6034239 * | Mar 6, 1997 | Mar 7, 2000 | Takeda Chemical Industries, Ltd. | Tricyclic compounds, their production and use |
| US20010039286 * | Feb 13, 2001 | Nov 8, 2001 | Kevin Dinnell | 2-aryl indole derivatives and their use as therapeutic agents |
| WO2006030739A1 | Sep 12, 2005 | Mar 23, 2006 | Takeda Pharmaceutical | Process for production of optically active amine derivatives |
| WO2008062468A2 | Oct 15, 2007 | May 29, 2008 | Cadila Healthcare Ltd | Process for the preparation of optically pure indeno [5,4-b] furan derivatives |
| WO2008106179A1 | Feb 26, 2008 | Sep 4, 2008 | Teva Pharma | Intermediates and processes for the synthesis of ramelteon |
| WO2009106966A1 | Feb 27, 2009 | Sep 3, 2009 | Medichem, S.A. | Process for preparing ramelteon. |
| WO2010055481A1 | Nov 12, 2009 | May 20, 2010 | Watson Pharma Private Limited | Process for the preparation of ramelteon |
| WO2010092107A1 * | Feb 11, 2010 | Aug 19, 2010 | Lek Pharmaceuticals D.D. | Synthesis of (s)-n-[2-(1,6,7,8-tetrahydro-2h-indeno-[5,4-b]furan-8-yl)ethyl]propionamide |
| WO2010103553A1 * | Mar 10, 2009 | Sep 16, 2010 | Industriale Chimica S.R.L. | Process for the preparation of ramelteon |
| EP0885210A1 | Mar 5, 1997 | Dec 23, 1998 | Takeda Chemical Industries, Ltd. | Tricyclic compounds, their production and use |
| US6034239 | Mar 6, 1997 | Mar 7, 2000 | Takeda Chemical Industries, Ltd. | Tricyclic compounds, their production and use |
| US20100152468 | Oct 16, 2009 | Jun 17, 2010 | Teva Pharmaceutical Industries Ltd. | Process for the synthesis of ramelteon and its intermediates |
| WO2008062468A2 * | Oct 15, 2007 | May 29, 2008 | Cadila Healthcare Ltd | Process for the preparation of optically pure indeno [5,4-b] furan derivatives |
| US5321154 * | Jul 31, 1992 | Jun 14, 1994 | Nagase & Company, Ltd. | Optical resolution of (.+-.)-2-(4-isobutylphenyl)-propionic acid |
| US6218429 * | May 10, 1999 | Apr 17, 2001 | Takeda Chemical Industries, Ltd. | Tricyclic compounds, their production and use |
| US6348485 * | Jun 8, 1999 | Feb 19, 2002 | Takeda Chemical Industries, Ltd. | Method for treating or preventing sleep disorders |
| US20080214559 * | Jan 7, 2008 | Sep 4, 2008 | Solvay Pharmaceuticals B.V. | Compounds with a combination of cannabinoid cb1 antagonism and serotonin reuptake inhibition |
| US20080242877 * | Feb 26, 2008 | Oct 2, 2008 | Vinod Kumar Kansal | Intermediates and processes for the synthesis of Ramelteon |
| WO2012035303A2 | Sep 16, 2011 | Mar 22, 2012 | Cipla Limited Et Al | A novel process for synthesis of ramelteon, and key intermediates for the synthesis of ramelteon |

Dedicated to lionel my son



My daughter Aishal

THEY KEEP ME GOING

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
http://anthonycrasto.jimdo.com/
blogs are

Congratulations! Your presentation titled “Anthony Crasto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
join my process development group on google
you can post articles and will be administered by me on the google group which is very popular across the world
LinkedIn group
blogs are
Subscribe to:
Posts (Atom)