Takeda’s flagship experimental cancer drug ixazomib is a giant leap closer to being filed with regulatory authorities around the globe for multiple myeloma, after turning in a solid performance in late-stage trials.
Takeda’s ixazomib soon to be filed for multiple myeloma
Read more at:
old paste cut paste
.gif)
CAS#: 1201902-80-8
Synonym: Ixazomib; MLN-9708.
IUPAC/Chemical name:
4-(carboxymethyl)-2-((R)-1-(2-(2,5-dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-1,3,2-dioxaborinane-4-carboxylic acid
CAMBRIDGE, Mass., May 23, 2013 – Takeda Pharmaceutical Company Limited (TSE:4502) today announced the initiation of an international phase 3 clinical trial evaluating once a week MLN9708 in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma who are not candidates for transplant. The multi-center study with MLN9708, an investigational, oral proteasome inhibitor, will be conducted in Europe and North America.———————-READ MORE AT
Description of Ixazomib: ixazomib is an orally bioavailable second generation proteasome inhibitor (PI) with potential antineoplastic activity. Ixazomib inhibits the activity of the proteasome, blocking the targeted proteolysis normally performed by the proteasome, which results in an accumulation of unwanted or misfolded proteins; disruption of various cell signaling pathways may follow, resulting in the induction of apoptosis. Compared to first generation PIs, second generation PIs may have an improved pharmacokinetic profile with increased potency and less toxicity. Proteasomes are large protease complexes that degrade unneeded or damaged proteins that have been ubiquinated
MLN9708 is an investigational proteasome inhibitor that, compared with bortezomib, has improved pharmacokinetics, pharmacodynamics, and antitumor activity in preclinical studies. MLN9708 rapidly hydrolyzes to MLN2238, the biologically active form. MLN9708 has a shorter proteasome dissociation half-life and improved pharmacokinetics, pharmacodynamics, and antitumor activity compared with bortezomib.MLN9708 has a larger blood volume distribution at steady state, and analysis of 20S proteasome inhibition and markers of the unfolded protein response confirmed that MLN9708 has greater pharmacodynamic effects in tissues than bortezomib. MLN9708 showed activity in both solid tumor and hematologic preclinical xenograft models, and we found a correlation between greater pharmacodynamic responses and improved antitumor activity. Moreover, antitumor activity was shown via multiple dosing routes, including oral gavage. Taken together, these data support the clinical development of MLN9708 for both hematologic and solid tumor indications. (source: Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16.).
| References |
1: Mullard A. Next-generation proteasome blockers promise safer cancer therapy. Nat Med. 2012 Jan 6;18(1):7. doi: 10.1038/nm0112-7a. PubMed PMID: 22227650.
2: Anderson KC. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. 2012 Feb 1;30(4):445-52. Epub 2012 Jan 3. PubMed PMID: 22215754.
3: Appel A. Drugs: More shots on target. Nature. 2011 Dec 14;480(7377):S40-2. doi: 10.1038/480S40a. PubMed PMID: 22169800.
4: Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, Li P, Hales P, Carsillo M, Neppalli VT, Berger AJ, Kupperman E, Manfredi M, Bolen JB, Van Ness B, Janz S. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res. 2011 Dec 1;17(23):7313-23. Epub 2011 Sep 8. PubMed PMID: 21903769.
5: Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, Richardson P, Anderson KC. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011 Aug 15;17(16):5311-21. doi: 10.1158/1078-0432.CCR-11-0476. Epub 2011 Jun 30. PubMed PMID: 21724551; PubMed Central PMCID: PMC3156932.
6: Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16. Erratum in: Cancer Res. 2010 May 1;70(9):3853. Hales, Paul [added]. PubMed PMID: 20160034.
7: Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010 Mar;15(5-6):243-9. Epub 2010 Jan 29. Review. PubMed PMID: 20116451.8: Marblestone JG. Ubiquitin Drug Discovery & Diagnostics 2009 – First Annual Conference. IDrugs. 2009 Dec;12(12):750-3. PubMed PMID: 19943215.
 Chemical structure of ixazomib
Chemical structure of ixazomib
Nasopharyngeal cancer is a sub-type of head and neck cancer that arises from the epithelial cells that cover the surface and line the nasopharynx. The incidence of nasopharyngeal cancer has been reported at approximately 0.5 to 2 new cases per year per 100,000 in Europe and the USA. Rottey et ah, Curr. Opin. Oncol., 23(3): 254-258 (201 1). There are three subtypes of nasopharyngeal cancer recognized in the World Health Organization (WHO) classification: (i) Type 1 – squamous cell carcinoma, typically found in the older adult population; (ii) Type 2 non-keratinizing carcinoma; and (iii) Type 3 – undifferentiated carcinoma. Treatment for nasopharyngeal cancer often involves radiotherapy and/or chemotherapy. There remains a continuing need for new and improved treatments for patients with nasopharyngeal cancer. There remains a further need to identify nasopharyngeal patients most likely to benefit from treatment with a proteasome inhibitor.
Proteasome inhibition represents an important new strategy in cancer treatment. King et al. , Science 274: 1652-1659 ( 1996), describes an essential role for the ubiquitin-proteasome pathway in regulating cell cycle, neoplastic growth and metastasis. The authors teach that a number of key regulatory proteins, including cyclins, and the cyclin-dependent kinases p21 and p27K,P ! , are temporally degraded during the cell cycle by the ubiquitin-proteasome pathway. The ordered degradation of these proteins is required for the cell to progress through the cell cycle and to undergo mitosis.
The proteasome inhibitor VELCADE© (bortezomib; N-2-pyrazinecarbonyl-L -phenylalanine -L- leucineboronic acid) is the first proteasome inhibitor to achieve regulatory approval. Mitsiades et ai, Current Drug Targets, 7: 1341 (2006), reviews the clinical studies leading to the approval of bortezomib for the treatment of multiple myeloma patients who have received at least one prior therapy. Fisher et ai , J. Clin. Oncol, 30:4867, describes an international multi-center Phase II study confirming the activity of bortezomib in patients with relapsed or refractory mantle cell lymphoma. Ishii et al, Anti-Cancer Agents in Medicinal Chemistry, 7:359 (2007), and Roccaro et al., Curr. Pharm. Biotech., 7: 1341 (2006), discuss a number of molecular mechanisms that may contribute to the antitumor activities of bortezomib. The proteasome inhibitorMLN9708 [2,2′-{2-[(lR)- l -( {[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3- methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid] is currently undergoing clinical evaluation for hematological and solid cancers. MLN9708 is a citrate ester which rapidly hydrolyzes to the active form [(lR)-l -({[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3-methylbutyl]boronic acid (MLN2238) on exposure to aqueous solution or plasma. MLN9708 has demonstrated anti-tumor activity in a range of hematological and solid tumor xenograft models (Kupperman et al. (2010) Cancer Res. 70: 1970- 1980),
Summary
The invention relates to the discovery that patients with nasopharyngeal cancer respond to treatment with MLN9708. In one aspect, the invention relates to the discovery of the increased expression of Nuclear Factor Kappa-B RelA 65,000 dalton subunit (NFKB p65) in biological samples comprising cells obtained from patients with nasopharyngeal cancer and responsive toMLN9708.
Accordingly, the invention features treating nasopharyngeal cancer patients withMLN9708 if a sample from the patient demonstrates an elevated expression of NFKB p65.
…………………………………..

or a pharmaceutically acceptable salt or a pharmaceutical composition or a boronic acid anhydride thereof.
[048| The compound of formula (II), [( l R)-l -( } [(2,5-dichlorobenzoyl)amino]acetyl} amino)-3- methylbutyljboronic acid (MLN2238) is disclosed in Olhava and Danca, U .S. Patent No. 7,442,830, herein incorporated by reference in its entirety. [049] In some other embodiments, Z and Z together form a moiety derived from a compound having at least two hydroxyl groups separated by at least two connecting atoms in a chain or ring, said chain or ring comprising carbon atoms and, optionally, a heteroatom orheteroatoms which can be N, S, or O, wherein the atom attached to boron in each case is an oxygen atom.
In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A) or (III-B):

(III-B), or a mixture thereof or a pharmaceutical composition thereof.
[054] In certain embodiments, wherein the alpha-hydroxy carboxylic acid orbeta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A):

or a pharmaceutical composition thereof.
[055] The compound of formula (III-A), 2,2′- {2-[( l i?)- l -( { [(2,5-dichlorobenzoyl)amino]acetyl } amino)- 3-methylbutyl]-5-oxo- l ,3,2-dioxaborolane-4,4-diyl} diacetic acid (MLN9708) is disclosed in Elliott et al. , WO 09/ 154737, herein incorporated by reference in its entirety
…………………………………………
Example 1: Synthesis of 4-(/?,S)-(carboxymethyl)-2-( (R)-I -(2-(2,5- dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-l,3,2-dioxaborinane-4- carboxylic acid (1-1)

Step l: 2,5-r(dichlorobenzoyI)aminolacetic acid
[0310] To a mixture of NaOH (12 g, 300 mmol) and glycine (18 g, 239 mmol) in water (120 mL) was added dropwise over 45 min a solution of 2,5-dichlorobenzoyl chloride (10 g, 48 mmol) in THF (15 mL) keeping the internal temperature below about 25 0C. After 1 h, the mixture was acidified with 2.0 M HCl (125 mL) keeping the internal temperature below about 5 0C. The resulting precipitate was collected by vacuum filtration. The crude product was recrystallized from water to give 2,5-[(dichlorobenzoyl)amino]acetic acid as a white, crystalline solid (6.1 g, 52%). mp 173.3 0C. 1H NMR (300 MHz, DMSOd6, δ): 12.72 (bs, IH), 8.89 (t, J = 6.0 Hz, IH), 7.54 (m, 2H), 7.48 (m, IH), 3.93 (d, J = 6.0 Hz). 13C NMR (75 MHz, DMSO-Ci6, δ): 41.6, 129.3, 129.6, 131.4, 132.2, 138.2, 171.4, 165.9. MS (ni/z): [M+H] calculated for C9H8Cl2NO3, 248.0; found, 248.0; [M+Na] calculated for C9H7Cl2NNaO3, 270.0; found 270.2.
[0311] 2,5-[(dichlorobenzoyl)amino]acetic acid was also be prepared via the following procedure: To a mixture of glycine (21.5 g, 286 mmol) in water (437 mL), was added 2.0 M NaOH (130 mL) and the resulting solution was cooled to 0 0C. A solution of 2,5-dichlorobenzoyl chloride (50.0 g, 239 mmol) in THF (75 mL) was added dropwise at such a rate that the internal temperature was maintained at 0 ± 1 0C. During the addition, the pH was controlled at 11.0 ± 0.2 using a pH controller titrated with 2.0 M NaOH. After complete addition, the mixture was stirred at 0 ± 1 0C for an additional 2 h. The mixture was then acidified with 2.0 M HCl (176 mL) to a final pH of 2.5. The resulting precipitate was collected by filtration, washed with cold water (125 mL), and dried at 45 0C in a vacuum oven to afford 2,5-[(dichlorobenzoyl)amino]acetic acid as a white solid (57.6 g, 97.3%). Step 2: 2,5-dichloro-N-f2-(( (lR’)-3-niethyl-l-r(3aS,4S.6S.7aR)-3a,5,5-trimethylhexahvdro-
4,6-methano-l,3,2-benzodioxaborol-2-yllbutyl }amino)-2-oxoethvπbenzamide
[0312] To a solution of 2,5-[(dichlorobenzoyl)amino]acetic acid (6.10 g, 24.6 mmol) and TBTU (8.34 g, 26.0 mmol) in DMF (40 mL) with an internal temperature below about 5 0C was added (IR)- 3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5-trimethylhexahydro-4,6-methano-l,3,2-benodioxaborol-2- yl]butan-l-amine»TFA (9.35 g, 24.7 mmol). DIPEA (13 mL, 75 mmol) was then added dropwise over 2 h keeping the internal temperature below about 5 0C. After 40 min, the mixture was diluted with EtOAc (90 mL), washed with 5% NaCl (150 mL), twice with 10% NaCl (2 x 40 mL), once with 2% K2CO3 (1 x 40 mL), once with 1% H3PO4 (1 x 40 mL), and once with 10% NaCl (1 x 40 mL). The resulting organic layer was concentrated to a thick oil, diluted with heptane (40 mL) and evaporated to yield 2,5-dichloro-N-[2-({ (lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l ,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide as a white solid which was used in the next step without purification.
Step 3: N,N\N’Wboroxin-2A6-triyltrisir(lR)-3-methylbutane-l J-diyllimino(2-oxoethane- 2,l-diyl)^ ^tris(2,5-dichlorobenzamide)
[0313] To a solution of 2,5-dichloro-N-[2-({(lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide (12.2 g, 24.6 mmol) in methanol/hexane (1 :1) (250 mL) were added IN HCl (30 mL, 30 mmol) and (2-methylpropyl)boronic acid (6.5 g, 64 mmol). The reaction mixture was allowed to stir overnight. The phases were separated and the methanol layer was washed twice with additional heptane (2 x 55 mL). The resulting organic layer was concentrated to about 10 mL and partitioned between 2.0M NaOH (30 mL) and DCM (25 mL). The DCM layer was washed once with additional 2.0M NaOH (5 mL). The basic aqueous layers were then combined, washed twice with DCM (2 x 25 mL) and acidified with IM HCl (60 mL). The resulting mixture was diluted with DCM (40 mL), the layers were separated, and the resulting aqueous layer was washed three times with DCM (3 x 10 mL). The combined DCM extracts were dried over MgSO4 (25 g) and evaporated to a thick oil. The product was precipitated with heptane (50 mL) and collected by filtration to yield N,N’,N”-{boroxin-2,4,6- -riyltris[[(lR)-3-methylbutane-l,l-diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) as a white solid (6.6 g, 74%). 1H NMR (300 MHz, DMSO-Cl6, δ): 8.93 (t, J – 6.0 Hz, IH), 8.68 (bs, IH), 7.63 (m, IH), 7.52 (m, 2H), 4.00 (d, J = 6.0 Hz, 2H), 2.62 (m, IH), 1.59 (m, IH), 1.33 (m, IH), 1.24 (m, IH), 0.81 (d, / = 5.9 Hz, 6H). 13C NMR (125 MHz, DMSO-Cl6, δ): 23.2, 25.8, 40.1, 40.7, 43.0, 129.0, 130.0, 131.0, 137.5, 165.0, 172.5. MS (m/z) in CH3CN: [M+H] calculated for C42H52B3Cl6N6O9, 1027.2; found, 1027.3; [M+Na] calculated for C42H51B3Cl6N6NaO9, 1049.2; found 1049.5.
Step 4: 4-(/?.S)-(carboxymethyl)-2-((/?)-l-(2-(2,5-dichlorobenzamido)acetamido)-3- methylbutyl)-6-oxo-l,3,2-dioxaborinane-4-carboxylic acid (1-1)
[0314] Form 1: To a solution of citric acid (2.75 g, 14.3 mmol) in EtOAc (85 mL) with an internal temperature of about 74 0C was added N,N’,N”-{boroxin-2,4,6-triyltris[[(lR)-3-methylbutane-l,l- diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) (5.00 g, 4.87 mmol) as a solid. The solution was cooled uncontrolled until the internal temperature was about 25 0C and the mixture was stirred overnight. The resulting precipitate was collected by filtration to yield 2,2′-{2-[(lR)-l-({ [(2,5- dichlorobenzoyl)amino]acetyl }amino)-3-methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid Form 1 as a crystalline solid (6.65 g, 88 %). 1H NMR (500 MHz, DMSOd6, δ 110 0C): 10.08 (s, IH), 8.69 (s, IH), 7.61 (s, IH), 7.52 (d, J = 1.3 Hz, 2H), 4.26 (d, J = 5.5 Hz, 2H), 2.70 (q, J = 14.5 Hz, 4H), 2.70 (bs, IH), 1.72 (sept, J – 6.5 Hz, IH), 1.42 (ddd, J = 5.2 Hz, J = 8.6 Hz, J = 13.9 Hz, IH), 1.28 (ddd, J = 5.3, J = 9.4 Hz, J = 14.3 Hz, IH), 0.91 (dd, J = 3.3 Hz, J = 6.6 Hz, 6H). MS (m/z) in CH3CN: [M+Na] calculated for C20H23BCl2N2NaO9, 539.1; found, 539.1.
Ixazomib citrate [USAN]
1,3,2-Dioxaborolane-4,4-diacetic acid, 2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo- [ACD/Index Name]
1,3,2-Dioxaborolane-4,4-diacetic acid,2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-
1239908-20-3 [RN]
2,2′-{2-[(1R)-1-{[N-(2,5-Dichlorbenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4,4-diyl}diessigsäure [German] [ACD/IUPAC Name]
2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacetic acid [ACD/IUPAC Name]
2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diacetic acid
2-[4-(carboxymethyl)-2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methyl-butyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
Acide 2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-méthylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacétique [French][ACD/IUPAC Name]
MLN9708









 InfoChem will be represented at the forthcoming ACS Meeting in San Diego. You will find Dr. Josef Eiblmaier, Dr. Valentina Eigner Pitto, and Dr. Peter Loew …
InfoChem will be represented at the forthcoming ACS Meeting in San Diego. You will find Dr. Josef Eiblmaier, Dr. Valentina Eigner Pitto, and Dr. Peter Loew …








 Historische Bilder der Landsberger Straße – An der Trambahnhaltestelle Holzapfelstraße endet – Münchner Straßen – München – Süddeutsche.de
Historische Bilder der Landsberger Straße – An der Trambahnhaltestelle Holzapfelstraße endet – Münchner Straßen – München – Süddeutsche.de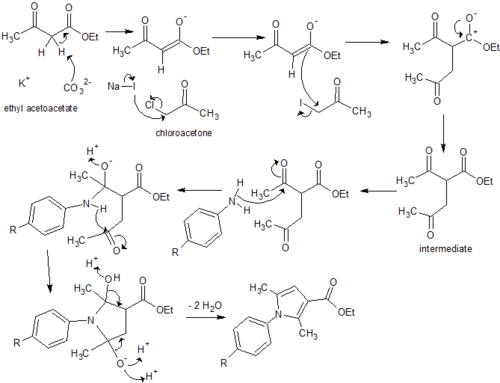








![Figure 9: Synthesis of ethyl 2,5-dimethyl-1-[p-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate](http://openwetware.org/wiki/Image:LMW_9-1.png)

