
Treating the flu?
They walked out together into the fine fall day, scuffling bright ragged leaves under their feet, turning their faces up to a generous sky really blue and spotless. At the first corner they waited for a funeral to pass, the mourners seated straight and firm as if proud in their sorrow. [...] “It seems to be a plague,” said Miranda, “something out of the Middle Ages. Did you ever see so many funerals, ever?”
— from “Pale Horse, Pale Rider” by Katherine Anne Porter (1939)
— from “Pale Horse, Pale Rider” by Katherine Anne Porter (1939)
And I looked, and behold a pale horse: and his name that sat on him was Death, and Hell followed with him.
— Revelations 6.8 (King James Version)
— Revelations 6.8 (King James Version)
 In 1918-1919 between 50 and 100 million people worldwide died from the flu. The "Spanish Flu" spread to nearly every part of the world with amazing speed, helped perhaps by the thousands of soldiers returning from Europe after the end of World War I. There was little that could be done to help the sick, and often people who were healthy one day were dead the next. The Spanish Flu was remarkable at the time in that it primarily killed young healthy adults, whereas most often it is very young children and elderly people who die from infectious disease.
In 1918-1919 between 50 and 100 million people worldwide died from the flu. The "Spanish Flu" spread to nearly every part of the world with amazing speed, helped perhaps by the thousands of soldiers returning from Europe after the end of World War I. There was little that could be done to help the sick, and often people who were healthy one day were dead the next. The Spanish Flu was remarkable at the time in that it primarily killed young healthy adults, whereas most often it is very young children and elderly people who die from infectious disease.
Oddly enough, after going through two successive waves of infection and mortality, the Spanish Flu pandemic disappeared almost abruptly. By the end of the 20th century, it was almost forgotten, and influenza had come to be regarded as one of the many childhood diseases that most people went through without much difficulty.
The situation today is quite different. Everyone is now highly sensitized to the threat of influenza. Stories about the so-called "Bird Flu" and now the "Swine Flu" have appeared regularly on television and in newspapers. Our society is more mobile than ever before, and we have seen examples of the rapid spread of diseases worldwide in recent years. Population is far more dense than it was in 1918, and diseases spread and mutate in crowded cities around the world far faster than ever before. People are deeply concerned about the possibility of a new influenza pandemic that could rival the Spanish Flu.
On the other hand, we also now know much more about how to prevent and how to treat illnesses like influenza. The best way to slow or stop the spread of influenza is through public health measures - simple things like frequent hand washing and avoiding contact with infected people. In addition, immunization is an important protective measure if a safe and effective vaccine can be developed.
But what about treating people who are already infected? Because influenza is a viral disease, antibiotics that can deal with bacterial infections will not work. The story of how drugs to treat serious cases of influenza were developed shows how structural biology, biochemistry and synthetic organic chemistry work hand-in-hand to produce new and useful chemical substances. It remains to be seen if they can help in the event of a pandemic outbreak, which many people think is a question of "when" rather than "if".
Treating the flu? Part 1: The Influenza Virus
 | Influenza is caused by RNA viruses of the family Orthomyxoviridae. These virions are roughly 80-120 microns in diameter. Their surfaces consist of a lipid bilayer derived from the membrane of the host cell, which is decorated by glycoproteins that project like spikes from the viral particle. About 80% of these spikes are hemagglutinin, a protein that facilitates binding the virion to a host cell. The remainder areneuraminidase, which is an enzyme that cleaves glycosidic linkages to the sugar neuraminic acid (also calledsialic acid). You have probably heard the different strains of the flu virus ("serotypes") referred to as "H1N1" or "H5N1". These names refer to the different subtypes of the two surface glycoproteins, differences that distinguish the serotypes immunogenically. |
There are several outstanding web sites that will tell you much more about the influenza virus. There is no point in just repeating what they contain here, so if you want more information you can follow the links below. Otherwise, click here to move to the next part of the drug development story.
- Some really nice electron micrographs of real influenza virions that show the hemagglutinin and neuraminidase "spikes".
- Influenza 101 - the virology blog. This takes you through the structure of the virus, how it enters cells, reproduces and then leaves to infect other cells. Very good quality information and very easy reading.
- Influenza Virus from Kimball's Biology Pages
- Influenza viruses on Wikipedia.
Treating the flu? Part 2: Targets for therapy
A drug must act by binding to and modulating the activity of some target receptor or enzyme. Viruses do not present very many potential targets because they typically have only a few unique proteins coded in their genomes. Recall that viruses hi-jack the enzymes of the host cell to manufacture new virions.
The Influenza A genome consists of 8 strands of RNA:
1. The HA gene. It encodes the hemagglutinin.
2. The NA gene. It encodes the neuraminidase.
3. The NP gene encodes the nucleoprotein. Influenza A, B, and C viruses have different nucleoproteins.
4. The M gene encodes two proteins (using different reading frames of the RNA): a matrix protein M1 and an ion channel M2 spanning the lipid bilayer.
5. The NS gene encodes two different non-structural proteins that are found in the cytoplasm of the infected cell but not within the virion itself.
6. – 8. one RNA molecule (PA, PB1, PB2) for each of the 3 subunits of the RNA polymerase.
2. The NA gene. It encodes the neuraminidase.
3. The NP gene encodes the nucleoprotein. Influenza A, B, and C viruses have different nucleoproteins.
4. The M gene encodes two proteins (using different reading frames of the RNA): a matrix protein M1 and an ion channel M2 spanning the lipid bilayer.
5. The NS gene encodes two different non-structural proteins that are found in the cytoplasm of the infected cell but not within the virion itself.
6. – 8. one RNA molecule (PA, PB1, PB2) for each of the 3 subunits of the RNA polymerase.
Drugs against Influenza A could potentially be developed to inhibit the activity of any of the products of the influenza genome, but in fact only drugs acting against the NA (neuraminidase) and the M2 (ion channel) proteins have been successfully developed to date.
The M2 inhibitors amantadine and rimantadine were the first effective drugs against influenza, but the M2 protein seems quite easy for the virus to modify so resistance rapidly develops against these drugs. The latest H1N1 virus that is causing pandemic concern is resistant to both amantadine and rimantadine. The drugs that are being used against current pandemic threat strains target the viral neuraminidase, and it is these that form the basis of our discussion on drug development.
Treating the flu? Part 3: Neuraminidase
This is only a very short description of this important enzyme. It assumes that you have some basic knowledge of what enzymes are and what they do. If you need more background information, your Biochemistry textbook or the Wikipedia article on enzymes are good places to start.
| Recall that the surface of the influenza virion is covered with spikes of hemagglutinin and neuraminidase. Hemagglutinin is a protein that binds tightly to the sugar portions of various cell-surface glycoproteins by recognizing and binding the sugarsialic acid, which is also called N-acetyl neuraminic acid. Sialic acid is found at the terminus of the carbohydrate portions of many cell-surface glycoproteins and plays a key role in cell-cell and cell-virus binding. The human ABO blood-group antigens are examples of sialylated oligosaccharides that play an important role in medical biochemistry. Hemagglutinin permits the influenza virus to attach to a host cell during the initial infection, which in turn causes the viral RNA to enter the cell by endocytosis. This is a common mechanism for infection and we know that many viruses including HIV as well as parasites such as the Plasmodium that causes malaria attack host cells via their cell-surface carbohydrates. However, the tight grip of viral hemagglutinin on cell-surface sialic acid is a problem when new viral particles need to break away from the host cell. |
The neuraminidase on the surface of the virion is necessary for new viral particles to break away from the host cell. Neuraminidase is a glycosidase (an enzyme that catalyzes the hydrolysis of glycosidic linkages) that specifically promotes the cleavage of sialic acid from glycoprotein saccharide chains. When the glycosidic linkage is cleaved by hydrolysis, the sialic acid falls off the cell surface. The viral particle is now no longer tethered to the host cell and can move off to infect other cells.
If the activity of neuraminidase is blocked, the new virions remain bound to the host cell and viral reproduction is prevented. You can view a Flash animation showing this concept here.

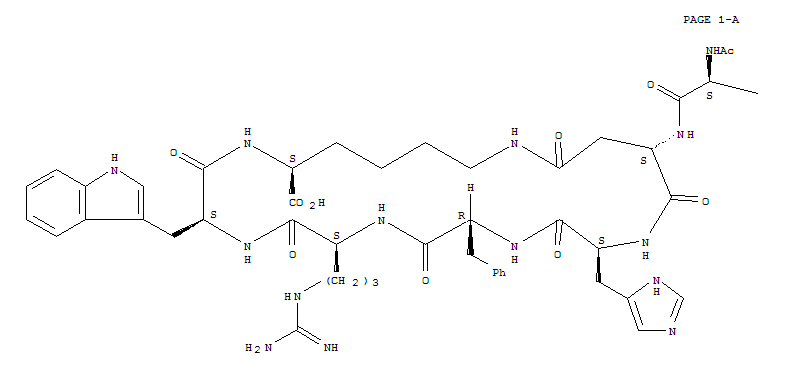
| The structure of the influenza A neuraminidase N9 bound to an analogue of sialic acid has been determined by X-ray crystallography, and a simplified ribbon diagram is shown here. The amino acid chains are represented by the yellow ribbons, and the bound inhibitor as well as some key side chain groups are shown in ball-and-stick format. The broad arrows designate regions in which the amino acid chains form a "beta sheet" structure, with the arrow heads indicating the C-terminal end of the sheet. Cylindrical sections represent "random coil" regions of the amino acid sequence. Notice that there is essentially no helical structure in this enzyme. This image shows only one sub-unit of the biologically active form of the enzyme which is actually a tetramer of identical sub-units. The binding site of the enzyme does not vary from strain to strain. It consists of 18 amino acid residues of which 12 are in direct contact with the bound sialic acid analogue (and presumably with sialic acid in catalytically active situations). Four of these 12 are positively-charged arginines, while another 4 are negatively-charged glutamic and aspartic acid residues. The remainder are neutral (tyrosine, asparagine, isoleucine and tryptophan). If you visit the RCSB Protein Data Bank you can find X-ray structures of many neuraminidases - this one is indexed under the code "1nna". The details of the structure are discussed in the original paper by Bossart-Whitaker et al. cited below.  |
Mark von Itzstein and coworkers (then at the Monash University Victorian College of Pharmacy in Melbourne Australia and now at the Institute for Glycomics at Australia's Griffith University) studied the mechanism of sialic acid hydrolysis catalyzed by influenza A N9 neuraminidase. This enzyme is what is called a retaining glycosidase because if the starting glycoside has the α-configuration (as shown) then the product that is formed will also have the α-configuration. In common with many glycosidase enzymes, its active site features a pair of carboxyl residues (Asp 151 and Glu 277 in the N9 neuraminidase they studied) which play central roles in the enzyme's catalytic mechanism. The proposed mechanism is shown below.

There are two important transition states shown in this mechanism, the first for the actual cleavage of the C-O bond leading to loss of the ROH fragment and the second for the formation of a new C-OH bond. In the first transition state, notice how the enzyme assists the ionization of a water molecule, the transfer of its proton to the leaving OR group, and stabilizes the transient positive charge on the ring oxygen.
With knowledge of how the enzyme functioned, von Itzstein decided that a compound that looked like the carbohydrate in that key first transition state would be a good candidate for an anti-influenza drug that would function by preventing the release of viral particles from infected cells. Click here to go to the next stage in the story - synthesizing and testing a new compound.
The story of how neuraminidase was identified as a target for anti-influenza drug development is briefly outlined by Graeme Laver, one of the key researchers in this field. You can read his March 2007 article in Education in Chemistry here.
Bossart-Whitaker, P.; Carson, M.; Babu, Y.S.; Smith, C.D.; Laver, W.G.; Air, G.M. J. Mol. Biol. 1993, 232, 1069–1083. (Link requires valid U of Manitoba Library ID).
von Itzstein, M. et al. Nature 1993, 363, 418-423. (Link requires valid U of Manitoba Library ID).
Treating the flu? Part 4: Developing Neuraminidase Inhibitors
Zanamivir (Relenza)
Note: this document should not be taken as any form of endorsement of the substances mentioned or as a recommendation for treatment.
With the information gained from structural and mechanistic studies on influenza A neuraminidase, von Itzstein and his team set out to devise and synthesize a stable molecule that looked sufficiently like the transition state to bind very tightly to the enzyme, thus inhibiting it. Recall that a transition state is not a stable isolable molecule, but it is possible to mimic the geometry of a proposed transition state with other chemical structures. These are called transition state analogues.
The proposed transition state for glycosidic bond cleavage in the mechanism previously outlined is shown here. Recall that for clarity the sugar structure has been simplified. It is evident that the reactive centre of the sugar ring is planar in this transition state. It is not possible to make a stable structure that has a double bond between position 2 and the ring oxygen similar to the partial double bond in the transition structure. Thus, von Itzstein et al. decided that a good inhibitor needed a double bond between positions 2 and 3 - that is, it should be a 2,3-dehydro derivative of sialic acid.
They also concluded that a strongly basic guanidino group should replace the hydroxyl at C-4 in the sialic acid structure. This would be positively charged at physiological pH and would bind strongly to a region of negative charge in the active site.

They synthesized and tested the structure shown in 1989 and found that it was indeed a potent and very selective inhibitor of influenza neuraminidase. Their synthetic route, published in the journal Carbohydrate Research in 1994, is shown below.

Although some of the reagents used in this synthesis may be unfamiliar, organic chemistry students should be able to recognize what is going on in each step. In the first step shown, the Lewis acid boron trifluoride etherate promotes an internal SN2 reaction in which the carbonyl of the acetamide displaces the acetate ester to form the new ring. Notice the inversion of configuration at C4. This is then subjected to another SN2 reaction in which the nucleophile is the azide anion N3-. The reagent is trimethylsilyl azide, which also provides mildly Lewis acidic activation for the displacement. Azide groups are excellent precursors for amines, and the reduction of the azide is easily carried out. You can see that some care must be taken here, since if the reaction is left too long the hydrogenation of the alkene will also occur. Simple alkaline hydrolysis removes the methyl ester and the acetate ester protecting groups, and then the amino group is converted into the desired guanidino function using formamidine sulfonic acid. This provided the desired neuraminidase inhibitor 4-deoxy-4-guanidino-2,3-dehydro-N-acetyl neuraminic acid, which ultimately has become the anti-influenza drug zanamivir (sold under the trade name Relenza by GlaxoSmithKline).
You can see how well zanamivir fits into the active site of influenza A neuraminidase from the X-ray crystal structure obtained by Zu et al. and indexed in theProtein Data Bank as 3b7e. This is an interesting structure because the enzyme is the neuraminidase from the A/Brevig Mission/1/1918 H1N1 strain, one of the viruses that caused the 1918 Spanish Flu. The genome of this virus was obtained from the frozen body of a woman who died in the Alaskan village of Brevig Missionin 1918. An interesting New York Times article describes the discovery of this virus (and incidentally the Johan Hultin who found the virus is no relation to Dr. Hultin!). It is another variation of the H1N1 strain that is at the centre of the 2009/2010 concern about Swine Flu.


The ribbon diagram has simplified the enzyme structure considerably - only those amino acids near the active site are shown, and only the most important ones that interact with zanamivir have their sidechains drawn. The drug molecule is shown in a space-filling representation in which oxygen is red, nitrogen is blue and carbon is white. Hydrogens are not shown. The diagram places the carboxylate group of zanamivir at the 6 o'clock position, while the guanidinium group is projecting backwards deep into the binding site. The hydroxylated sidechain is projecting forward at about 9 o'clock. The schematic drawing (based on a diagram from the book by Levy and Fugedi referenced below) shows the key contacts between the enzyme and the drug.
Numerous other synthetic routes to zanamivir have been published since the original synthesis shown here, and you can be very sure that the industrial synthesis isquite different. The problem with Zanamivir is that it cannot be administered orally. Because the guanidino group is strongly basic, if it were taken orally it would be protonated in the stomach. The resulting positively-charged structure could not be taken up from the gut. Zanamivir is usually administered by inhalation, but this is not as acceptable to many people as a pill would be, and does not give a particularly high level of bioavailability.
Given this problem with zanamivir, it is not surprising that others tried to find similar compounds to inhibit influenza neuraminidase that could be orally administered. Click here to find out about the second-generation drug oseltamivir (Tamiflu).
von Itzstein, M.; Wu, W.-Y.; Jin, B. Carbohydrate Research 1994, 259, 301-305.
Taylor, N.R.; von Itzstein, M. J. Med. Chem. 1994, 37, 616–624.
Magano, J. Chem. Rev. 2009, in press. (You must have a valid U of Manitoba library ID to access the full-text article)
Xu, X.; Zhu, X.; Dwek, R.A.; Stevens, J.; Wilson, I.A. J.Virol. 2008, 82, 10493-10501.
Treating the flu? Part 4: Developing Neuraminidase Inhibitors
Other neuraminidase inhibitors
Research and development of new anti-influenza drugs has not stopped. The need for more effective drugs remains a powerful incentive for academic and industrial scientists, and there is of course a strong profit motive as well.

One compound that is now in clinical trials is peramivir, under development by BioCryst Pharmaceuticals. If you look at the structure of peramivir, you can see its family resemblance to other neuraminidase inhibitors. However, peramivir must be administered by injection because it has rather poor oral bioavailability. In fact, peramivir was initially developed by Johnson and Johnson but was abandoned because it was not orally active. Renewed interest in it as an injectable drug may be because only the most severe cases of influenza really need antiviral therapy, and such patients are likely already hospitallized.
Another new compound is CS-8958, from Japan's Daiichi Sankyo Co. Ltd. This compound is structurally very similar to zanamivir, differing only in the functionalization of the hydroxylated sidechain.
CS-8958 is a prodrug and not the active form. The octyl ester group is hydrolyzed in the liver, releasing the active neuraminidase inhibitor which only differs from zanamivir in having a methyl ether at the C7 position rather than a hydroxyl group. The main advantage of CS-8958 is that it is long-acting. Oseltamivir and zanamivir must be taken twice daily, but in a clinical study a single inhaled treatment with CS-8958 gave the same anti-influenza effect as twice-daily doses of oseltamivir over 5 days.















 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..


 Location in Madhya Pradesh
Location in Madhya Pradesh














 LATUR AIRPORT
LATUR AIRPORT











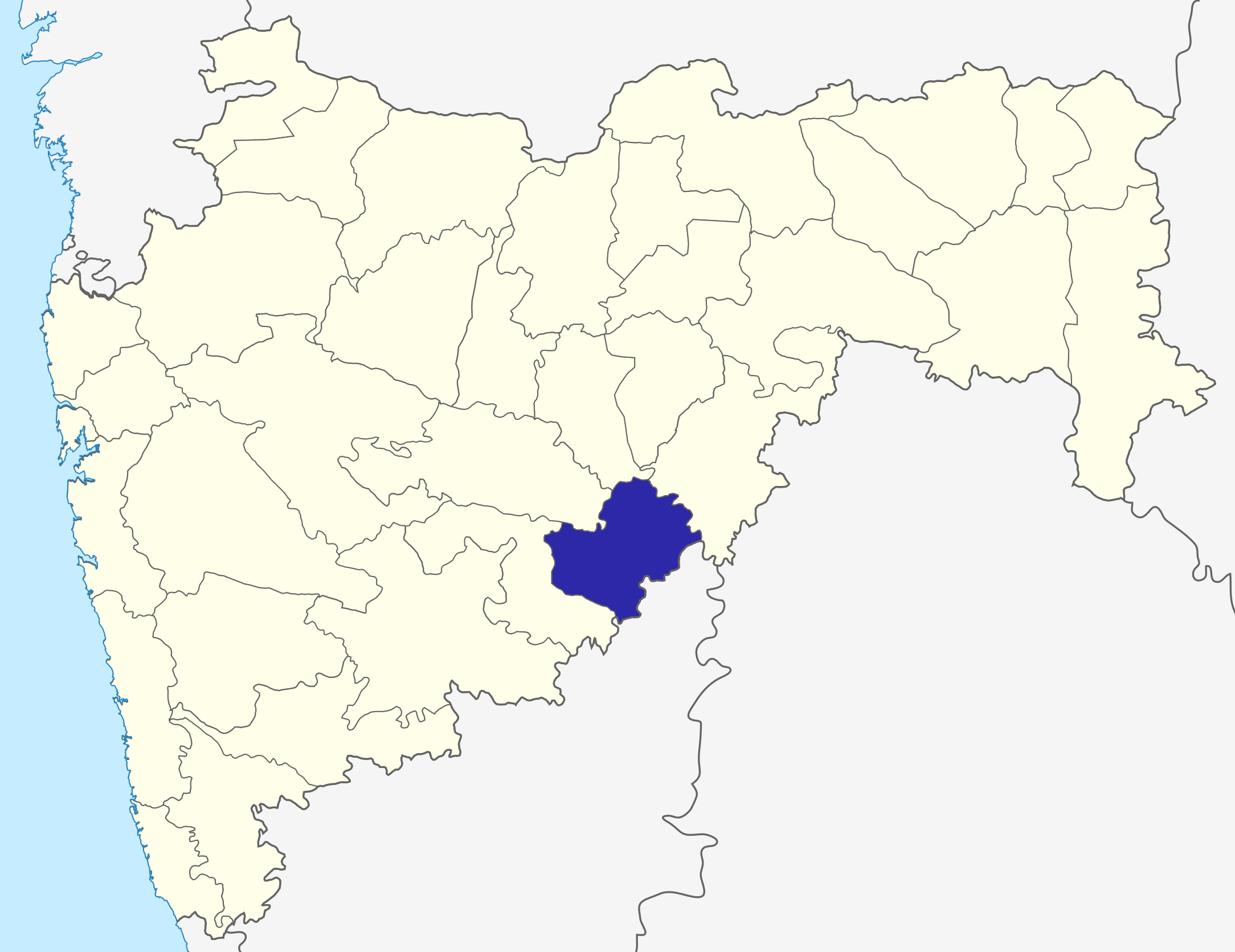
 The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the