
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

Synthetic Communications: An International Journal for Rapid Communication of Synthetic Organic Chemistry
A practical and economical approach to synthesize sitagliptin
Volume 43, Issue 24, 2013
- DOI:
- 10.1080/00397911.2013.773353
pages 3281-3286
1Kuaile Lin, Zhengyan Cai, Weicheng Zhou*
State Key Lab of New Drug & Pharmaceutical Process, Shanghai Key Lab of
Anti-Infectives,
Shanghai Institute of Pharmaceutical Industry, State Institute
ofPharmaceutical Industry, Shanghai 200437, China
* Corresponding author: Weicheng Zhou, profzhouwc@yahoo.com.cn, Tel./fax: +8621 35052484
Economic syntheses of sitagliptin phosphate monohydrate, acknowledged as the first dipeptidyl peptidase 4 (DPP-4) inhibitor, have been achieved in an overall yield of 42.4% in four steps from 1-{3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl}-4-(2,4,5-trifluorophenyl)butane-1,3-dione. The key stereoselective reduction of this process was carried out by NaBH4/HCOOH instead of expensive and toxic catalysts or ligands.
LOOK FOR SUPPLEMENTARY INFO IN ABOVE PAPER


1H NMR SITAGLIPTIN PHOSPHATE MONOHYDRATE



tga
 .
P.S. : The
views expressed are my personal and in no-way suggest the views of the
professional body or the company that I represent.
.
P.S. : The
views expressed are my personal and in no-way suggest the views of the
professional body or the company that I represent.

...............................
NMR SEE AN ONLINE NMR BELOW
NMR............http://file.selleckchem.com/downloads/nmr/S400205-Sitagliptin-phosphate-monohydrate-HNMR-Selleck.pdf
......................
PAPER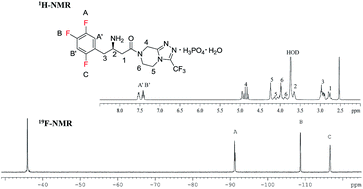
http://pubs.rsc.org/en/content/articlelanding/2014/an/c4an01681e#!divAbstract
CHECK OUT PREDICTIONS UNDERSTAND THE SIGNALS PREDICTIONS 1H NMR P.S. : The
views expressed are my personal and in no-way suggest the views of the
professional body or the company that I represent.
P.S. : The
views expressed are my personal and in no-way suggest the views of the
professional body or the company that I represent.
PREDICTIONS 13 C NMR LOOK FOR DELTA VALUES OF GROUPS
COSY NMR PREDICTION
.png)
BELOW PAPENT DESCIBES THIS DRUG WELL IS RANDOMLY CHOSEN
http://www.google.com/patents/EP2491040A2?cl=en


 Further, WO 2005/097733 discloses a method for preparing sitagliptin by stereoselectively reducing an enamine employing a rhodium-based catalyst, [Rh(cod)Cl]2 having a chiral diphosphine ligand, as shown in Reaction Scheme 4.
Further, WO 2005/097733 discloses a method for preparing sitagliptin by stereoselectively reducing an enamine employing a rhodium-based catalyst, [Rh(cod)Cl]2 having a chiral diphosphine ligand, as shown in Reaction Scheme 4.









LOOK FOR SUPPLEMENTARY INFO IN ABOVE PAPER
1H NMR SITAGLIPTIN PHOSPHATE MONOHYDRATE
tga
.
P.S. : The
views expressed are my personal and in no-way suggest the views of the
professional body or the company that I represent.
...............................
NMR SEE AN ONLINE NMR BELOW
NMR............http://file.selleckchem.com/downloads/nmr/S400205-Sitagliptin-phosphate-monohydrate-HNMR-Selleck.pdf
......................
PAPER
http://pubs.rsc.org/en/content/articlelanding/2014/an/c4an01681e#!divAbstract
Quantitative analysis of sitagliptin using the 19F-NMR method: a universal technique for fluorinated compound detection
*
Corresponding authors
a
State Key Laboratory of
Natural Medicines, Department of Pharmaceutical Analysis, China
Pharmaceutical University, Nanjing 210009, China E-mail:
ayanju@163.com
b
Shanghai Institute for Food and Drug Control, Shanghai 201203, China
c
Department of Chemistry
and Chemical Engineering, Royal Military College of Canada, Kingston,
Canada
d
Pharmaceutical Research
Institute, China Pharmaceutical University, Nanjing 210009, China E-mail:
cpunmrswb@163.com
Analyst, 2015,140, 280-286
DOI:
10.1039/C4AN01681E
 Vishva Shah, Royal Military College of Canada
Vishva Shah, Royal Military College of CanadaCHECK OUT PREDICTIONS UNDERSTAND THE SIGNALS PREDICTIONS 1H NMR
P.S. : The
views expressed are my personal and in no-way suggest the views of the
professional body or the company that I represent.PREDICTIONS 13 C NMR LOOK FOR DELTA VALUES OF GROUPS
COSY NMR PREDICTION
BELOW PAPENT DESCIBES THIS DRUG WELL IS RANDOMLY CHOSEN
http://www.google.com/patents/EP2491040A2?cl=en
The present invention relates to a novel method of preparing sitagliptin, and intermediates used therein. BACKGROUND OF THE INVENTION
Sitagliptin phosphate is a selective inhibitor of the second generation dipeptidyl peptidase IV (DPP-4) and used to maintain the systemic concentration of incretin hormone at an optimum level. Sitagliptin phosphate monohydrate was approved in October 2006 by the US Food and Drug Administration (FDA) as an adjuvant in dietetics or kinesiatrics for treatment of patients with type-2 diabetes and it is marketed in the United States and Korea under the trade name of JANUVIA™ (as a single agent).
Various methods for preparing sitagliptin and sitagliptin phosphate have been developed. For example, International Patent Publication WO 2003/004498 discloses a method of introducing a chiral-amine group using a chiral pyrazine derivative and to prepare sitagliptin by Arndt-Eistert Homologation using t-butoxylcarbonylamino-4-(2,4,5-trifluorophenyl)-butyric acid as a sitagliptin intermediate, as shown in Reaction Scheme 1.
Reaction Scheme 1
Wherein,
Boc is tert-butoxycarbonyl, TEA is trimethylamine, HOBt is 1- hydroxybenzotriazole, EDC is N-ethyl-N'-(3- dimethylaminopropyl)carbodiimide, and DIPEA is N,N-diisopropylethylamine.
International Patent Publication WO 2004/087650 discloses a method for preparing sitagliptin phosphate comprising the steps of: subjecting (2,4,5- trifluorophenyl)acetic acid to two-step reactions to obtain methyl 4-(2,4,5- trifluorophenyl)-3-oxophenylbutylate; conducting a stereoselective reduction of the resulting compound in the presence of (S)-BrNAP-RuCl2-Et3N under a high hydrogen pressure; hydrolyzing the reduced product to obtain (3S)-3-hydroxy- 4-(2,4,5-trifluorophenyl)-butyric acid, a key sitagliptin intermediate; and subjecting (3S)-3-hydroxy-4-(2,4,5-trifluorophenyl)-butyric acid to seven-step processes to obtain sitagliptin phosphate, as shown in Reaction Scheme 2.
Reaction Scheme 2
Wherein,
BINAP is 2,2'-bis(diphenylphosphino)-l,l'-binaphthyl, EDC is N-ethyl-N'-(3- dimethylaminopropyl)carbodiimide, Bn is benzyl, DIAD is diisopropyl azodicarboxylate, NMM is N-methylmorpholine, and ACN is acetonitrile.
Further, International Patent Publication WO 2004/085661 discloses a method for preparing sitagliptin by stereoselectively reducing an enamine using a platinum catalyst, PtO2, as shown in Reaction Scheme 3. Reaction Scheme 3
The document [J. Am. Chem. Soc, 2009, 131, p.l 1316-11317] discloses a method for preparing sitagliptin by stereoselectively reducing an enamine using a ruthenium-based catalyst, Ru(OAc)2 having a chiral diphosphine ligand, and International Patent Publication WO 2009/064476 discloses a method for preparing sitagliptin by stereoselectively reducing an enamine using Ru(OAc)2and a chiral diphosphine ligand, or using a chiral acid together with a borohydride reducing agent (e.g., NaBH4).
Reaction Scheme 5
Example 1: Preparation of (2S)-2-(2,4,5-trifluorobenzyl)- oxirane
Step 1 : Preparation of (2S)-3-(2A5-trifluorophenyl)-l-chloro-2-propanol
Magnesium (Mg) (1.26 g) was suspended in tetrahydrofuran (THF) (10 ml), and a drop of 1,2-dibromoethane was added thereto. To the resulting mixture, 2,4,5-trifluorobenzene bromide (0.55 g) was added dropwise slowly and then stirred for 30 min. 2,4,5-trifluorobenzene bromide (9.0 g) dissolved in THF (50 ml) was added slowly dropwise to the resulting mixture for 30 min and then stirred at room temperature for 1 hour. Cul (0.72 g) was added to the resulting mixture and the reaction temperature was cooled to 0°C . (S)- epichlorohydrin (4.1 ml) dissolved in THF (40 ml) was added dropwise to the resulting mixture slowly over 30 min, heated to room temperature, and stirred for 2 hours. Satuated NH4CI (50 ml) and ethyl acetate (50 ml) were added to the resulting mixture, and the organic layer formed thereafter was separated. The separated organic layer was washed with 50 ml of satuated saline, dried over MgSO4, and filtered. The organic solvent was removed from the filtrate under a reduced pressure to obtain the title compound.
Step 2: Preparation of (2S)-2-(2,4,5-trifluorobenzyl)-oxirane
(2S)-3-(2,4,5-trifluorophenyl)-l-chloro-2-propanol obtained in step 1 was dissolved in methanol (50 ml), and NaOH (2.3 g) was added dropwise thereto. The resulting mixture was stirred for 1 hour and methanol was removed therefrom under a reduced pressure. Water (50 ml) and ethyl acetate (50 ml) were added to the resulting mixture, and the organic layer formed thereafter was separated. The separated organic layer was washed with satuated saline, dried over MgSO4, and filtered to remove MgSO4. The organic solvent was removed from the filtrate under a reduced pressure to obtain the title compound (6.8 g; yield: 80%).
1H-NMR(300MHz, CDC13): 6 7.17-7.05 (2H, m), 6.96-6.88 (2H, m), 3.16-3.13 (1H, m) 3.14 (1H, dd, J=4.68, 14.7), 2.82-2.77 (2H, m), 2.54-2.47 (1H, m). Preparation Example 2: Preparation of (2S)-2-(2,4,5-trifIuorobenzyl)- oxirane
Step 1 : Preparation of (2S)-3-(2A5-trifluorophenyl)-l-chloro-2-propanol
2N -PrMgCl (26 ml) suspended in THF was added dripwise to the 2,4,5-trifluorobenzene bromide (9.55 g) dissolved in THF (30 ml) at -15 °C for 60 min. Cul (0.72 g) was added thereto at -15 °C , and heated to -10 °C . (S)- epichlorohydrin (4.1 ml) dissolved in THF (40 ml) was added slowly to the resulting mixture, and stirred at 0 °C for 1 hour. Satuated NH4C1 (50 ml) and ethyl acetate (50 ml) were added to the resulting mixture, and the organic layer formed thereafter was separated. The separated organic layer was washed with 50 ml of satuated saline, dried over MgSO4, and filtered to remove MgSO4. The organic solvent was removed from the filtrate under a reduced pressure to obtain the title compound.
Step 2: Preparation of (2S)-2-(2,4,5-trifluorobenzyl)-oxirane (2S)-3-(2,4,5-trifluorophenyl)-l-chloro-2-propanol obtained in step 1 was dissolved in 50 ml of methanol, and NaOH (2.3 g) was added dropwise thereto. A mixture was stirred for 1 hour, and methanol was removed therefrom under a reduced pressure. Water (50 ml) and ethyl acetate (50 ml) were added thereto, and the organic layer formed thereafter was separated. The separated organic layer was washed with satuated saline, dried over MgSO4, and filtered to remove MgSO4. The organic solvent was removed from the filtrate under a reduced pressure to obtain the title compound (7.6 g; yield: 85%). Example 1: Preparation of Sitagliptin
Step 1: Preparation of (2R)-l-(2,4,5-trifluorophenyl -4-pentene-2-ol
CuBr(CH3)2 (3.3 g) was added to a reactor under the nitrogen atmosphere and cooled to -78 °C . Vinylmagnesium bromide (240 ml) was added slowly to the reactor and stirred for 20 min. (2S)-2-(2,4,5- trifluorobenzyl)-oxirane (30 g) dissolved in THF (90 ml) was added dropwise slowly over 30 min, stirred at -78 °C for 30 min, and heated to 0 °C . 2N aqueous HC1 (300 ml) was added slowly to the resulting mixture, and the organic layer formed thereafter was separated. The separated organic layer was washed twice with satuated saline, dried over MgSO4, and filtered. The organic solvent was removed from the filtrate under a reduced pressure to obtain the title compound (34.5 g; yield: 100%).
1H-NMR(300MHz, CDC13): δ 7.15-7.06 (1H, m), 6.94-6.86 (1H, m), 5.85-5.79 (1H, m), 5.20-5.14 (2H, m), 3.90-3.85 (1H, m), 3.82 (1H, dd, J=4.6, 18.5), 2.69 (1H, dd, J=7.9, 14.0), 2.37-2.32 (1H, m), 2.24-2.17 (1H, m), 1.86(1H, Br). Step 2: Preparation of (2S)-l-(2-azido-4-pentenyl)-2A5-trifluorobenezene
Dichloromethane (300 ml) was added to the (2R)-1 -(2,4,5- trifluorophenyl)-4-pentene-2-ol obtained in step 1, and cooled to 0°C . Triethylamine (20.4 ml) and 4-dimethylaminopyridine (DMAP) (1.57 g) were added successively to the mixture, and methansulfonyl chloride (1 1.2 ml) was added dropwise thereto for 30 min. The resulting mixture was stirred for 1 hour, water (150 ml) was added, and the organic layer formed thereafter was separated. The separated organic layer was washed twice with satuated saline, dried over MgSO4, and filtered. The organic solvent was removed from the filtrate under a reduced pressure. The residue thus obtained was dissolved in DMF (300 ml), and NaN3 (9.91 g) was added thereto. The resulting mixture was heated to 70 °C , stirred for 2 hours, and cooled to room temperature. And then water (150 ml) and ethyl acetate (150 ml) were added to the resulting mixture, and the organic layer formed thereafter was separated. The organic layer was washed twice with 150 ml of satuated saline, dried over MgSO4, and filtered. The organic solvent was removed from the filtrate under a reduced pressure to obtain the title compound (31.5 g; yield: 94%).
1H-NMR(300MHz, CDC13): δ 7.11-7.02 (1H, m), 7.97-6.87 (1H, m), 5.89-5.80 (1H, m), 5.23-5.17 (1H, m), 3.63-3.59 (1H, m), 2.87 (1H, dd, J=4.7, 18.7), 2.68 (1H, dd, J=7.9, 13.7), 2.38-2.17 (2H, m). Step 3: Preparation of (3R)-3-azido-4-(2A5-trifluorophenyl)-butyric acid
Acetonitril (300 ml) and water (300 ml) were added to the (2S)-l-(2- azido-4-pentenyl)-2,4,5-trifluorobenezene obtained in step 2, and cooled to 0°C . RuCl3 (0.5 g) and NaIO4 (93 g) were added to the mixture successively, and stirred for 5 hours. Ethyl acetate (90 ml) was added to the resulting mixture, filtered and the organic layer formed thereafter was separated. The separated organic layer was washed with IN HC1 (300 ml), satuated aqueous Na2S2O3 (300 ml) and satuated saline (300 ml), successively, dried over MgSO4, and filtered. The organic solvent was removed from the filtrate under a reduced pressure to obtain the title compound (32.2 g; yield: 100%). 1H-NMR(300MHz, CDC13): 5 10.5 (1H, br), 7.17-7.05 (1H, m), 7.02-
6.87 (1H, m), 4.14-4.03 (1H, m), 2.94-2.78 (2H, m), 2.65-2.51 (2H, m).
Step 4: Preparation of (3R)-3-azido-l-(3-trifluoromethyl-5,6-dihydro-8H- [ 1 ,2,41triazolor4,3-alpyrazin-7-yl)-4-(2,4,5-trifluorophenyl)-butan- 1 -one
(3R)-3-azido-4-(2,4,5-trifluorophenyl)-buryric acid (5 g) obtained in step 3 and triazole derivative of formula (VI) (5.3 g) were added to DMF (40 ml) and water (20 ml), stirred for 15 min, and cooled to 10°C . N- methylmorpholine (2.4 ml) was added to the mixture, stirred for 10 min, and cooled to 0 °C . EDC (5.6 g) was added to the resulting mixture, and stirred for 1 hour. Ethyl acetate (50 ml) and water (25 ml) were added to the resulting mixture, and the organic layer formed thereafter was separated. The separated organic layer was washed four times with 50 ml of satuated saline, dried over MgSO4, and filtered. The organic solvent was removed from the filtrate under a reduced pressure to obtain the title compound (7.8 g; yield: 93%).
1H-NMR(300MHz, CDC13): δ 7.20-7.11 (1H, m), 6.99-6.90 (1H, m), 5.20-4.96 (2H, m), 4.28-4.05 (5H, m), 2.98-2.67 (4H, m).
Step 5: Preparation of sitagliptin
(3R)-3-azido- 1 -(3-trifluoromethyl-5,6-dihydro-8H-[ 1 ,2,4]triazolo[4,3- a]pyrazin-7-yl)-4-(2,4,5-trifluorophenyl)-butan-l-one (6.4 g) obtained in step 4 and triphenylphosphin (4.3 g) were dissolved in THF (74 ml), heated to 50 °C , and stirred for 2 hours. An aqueous NH4OH (37 ml) was added to the resulting mixture and stirred for 10 hours. THF was removed from the resulting mixture under a reduced pressure, HCl (30 ml) and ethyl acetate (60 ml) were added threreto, and stirred. The water layer separated from the mixture was washed twice with 30 ml of n-hexane, satuated sodium bicarbonate was added to the water layer, and extracted three times with 60 ml of ethyl acetate. The resulting extracts were dried over MgSO4, and filtered. The organic solvent was removed from the filtrate under a reduced pressure to obtain the title compound (5.2 g; yield: 86%).
1H-NMR(300MHz, CDC13): δ 7.14-7.06 (1H, m), 7.00-6.88 (1H, m), 5.13-4.88 (2H, m), 4.24-3.80 (4H, m), 3.58 (1H, m), 2.85-2.66 (2H, m), 2.61-2.46 (2H, m), 2.11 (3H, br).
ABOVE IS ONLY ONE EXAMPLE LEADING TO SITAGLIPTIN

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
 .
.

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus
 amcrasto@gmail.com
amcrasto@gmail.com
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus Googleplus
Googleplus
amcrasto@gmail.com
.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.