
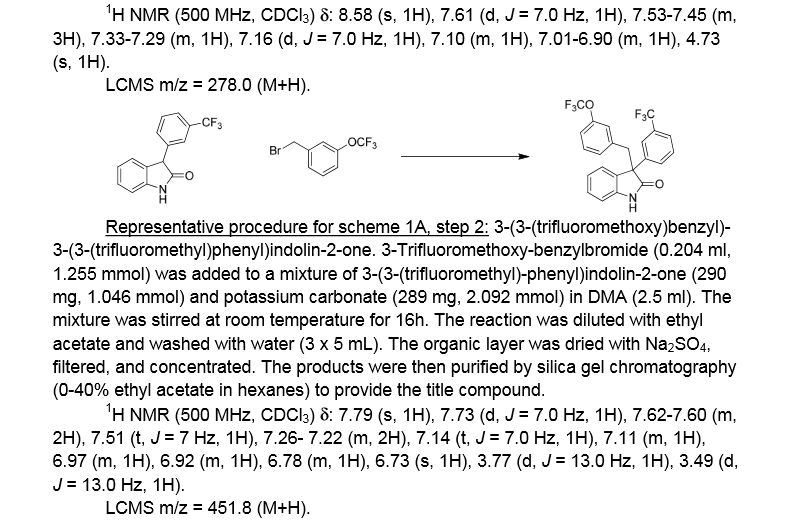
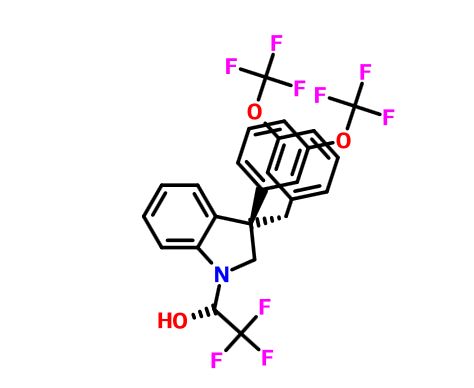
Elotuzumab
Approved nov 30 2012
A SLAMF7-directed immunostimulatory antibody used to treat multiple myeloma.
(Empliciti®)
HuLuc-63;BMS-901608
cas 915296-00-3


Elotuzumab (brand name Empliciti, previously known as HuLuc63) is a humanized monoclonal antibody used in relapsed multiple myeloma.[1] The package insert denotes its mechanism as a SLAMF7-directed (also known as CD 319) immunostimulatory antibody.[2]
Approvals and indications
In May 2014, it was granted “Breakthrough Therapy” designation by the FDA. [3] On November 30, 2015, FDA approved elotuzumab as a treatment for patients with multiple myeloma who have received one to three prior medications.[1] Elotuzumab was labeled for use with lenalidomide and dexamethasone. Each intravenous injection of elotuzumab should be premedicated with dexamethasone, diphenhydramine, ranitidine and acetaminophen.[2]Elotuzumab is APPROVED for safety and efficacy in combination with lenalidomide and dexamethasone.
Monoclonal antibody therapy for multiple myeloma, a malignancy of plasma cells, was not very clinically efficacious until the development of cell surface glycoprotein CS1 targeting humanized immunoglobulin G1 monoclonal antibody – Elotuzumab. Elotuzumab is currently APPROVED in relapsed multiple myeloma.
Elotuzumab (HuLuc63) binds to CS1 antigens, highly expressed by multiple myeloma cells but minimally present on normal cells. The binding of elotuzumab to CS1 triggers antibody dependent cellular cytotoxicity in tumor cells expressing CS1. CS1 is a cell surface glycoprotein that belongs to the CD2 subset of immunoglobulin superfamily (IgSF). Preclinical studies showed that elotuzumab initiates cell lysis at high rates. The action of elotuzumab was found to be enhanced when multiple myeloma cells were pretreated with sub-therapeutic doses of lenalidomide and bortezomib. The impressive preclinical findings prompted investigation and analysis of elotuzumab in phase I and phase II studies in combination with lenalidomide and bortezomib.
Elotuzumab As Part of Combination Therapy: Clinical Trial Results
Elotuzumab showed manageable side effect profile and was well tolerated in a population of relapsed/refractory multiple myeloma patients, when treated with intravenous elotuzumab as single agent therapy. Lets’ take a look at how elotuzumab fared in combination therapy trials,
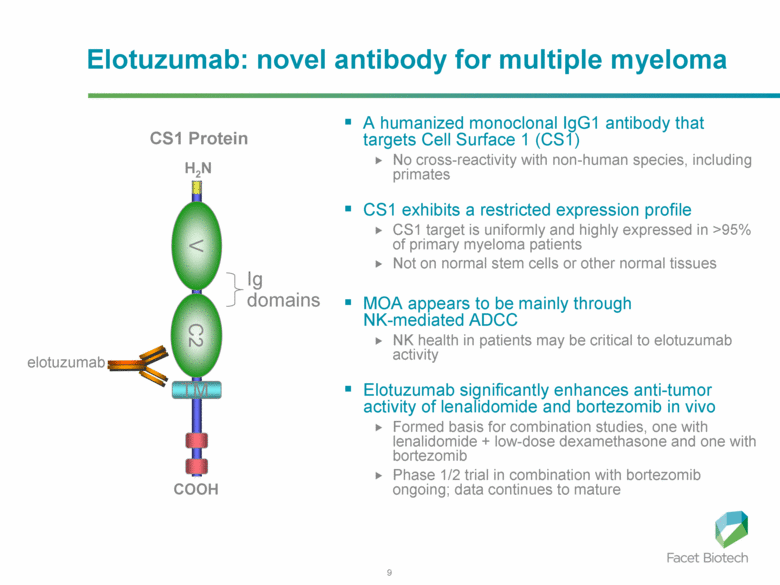
In phase I trial of elotuzumab in combination with Velcade/bortezomib in patients with relapsed/refractory myeloma, the overall response rate was 48% and activity was observed in patients whose disease had stopped responding to Velcade previously. The trial results found that elotuzumab enhanced Velcade activity.
A phase I/II trial in combination with lenalidomide and dexamethasone in refractory/relapsed multiple myeloma patients showed that 82% of patients responded to treatment with a partial response or better and 12% of patients showed complete response. Patients who had received only one prior therapy showed 91% response rate with elotuzumab in combination with lenalidomide and dexamethasone.
Phase I/II trials of the antibody drug has been very impressive and the drug is currently into Phase III trials. Two phase III trials are investigating whether addition of elotuzumab with Revlimid and low dose dexamethasone would increase the time to disease progression. Another phase III trial (ELOQUENT 2) is investigating and comparing safety and efficacy of lenalidomide plus low dose dexamethasone with or without 10mg/kg of elotuzumab in patients with relapsed/refractory multiple myeloma.
Elotuzumab is being investigated in many other trials too. It is being evaluated in combination with Revlimid and low-dose dexamethasone in multiple myeloma patients with various levels of kidney functions, while another phase II study is investigating elotuzumab’s efficacy in patients with high-risk smoldering myeloma.
The main target of multiple myeloma drug development is to satisfy the unmet need for drugs that would improve survival rates. Elotuzumab is an example that mandates much interest in this area and should be followed with diligence.

On November 30, 2015, the U. S. Food and Drug Administration
approved elotuzumab (EMPLICITI, Bristol-Myers Squibb Company) in
combination with lenalidomide and dexamethasone for the treatment of
patients with multiple myeloma who have received one to three prior
therapies.
Elotuzumab is a monoclonal antibody directed against Signaling
Lymphocyte Activation Molecule Family 7 (SLAMF7). SLAMF7 is present on
myeloma cells and is also present on natural killer cells.
The approval was based on a multicenter, randomized, open-label,
controlled trial evaluating progression-free survival (PFS) and overall
response rate (ORR) in patients with relapsed or refractory multiple
myeloma who had received 1 to 3 prior lines of therapy. A total of 646
patients were randomized (1:1) to receive elotuzumab in combination with
lenalidomide and dexamethasone (n=321) or lenalidomide plus
dexamethasone alone (n=325). Patients continued treatment until disease
progression or the development of unacceptable toxicity.
The trial demonstrated a statistically significant improvement in
both PFS and ORR, the trial’s co-primary endpoints. The median PFS in
the elotuzumab-containing arm was 19.4 months and 14.9 months in the
lenalidomide plus dexamethasone alone arm (hazard ratio 0.70, 95% CI:
0.57, 0.85; p = 0.0004). The ORR in the elotuzumab-containing arm was
78.5% (95% CI: 73.6, 82.9) compared to 65.5% (95% CI: 60.1, 70.7) in the
lenalidomide plus dexamethasone alone arm (p=0.0002).
The safety data reflect exposure in 318 patients to elotuzumab in
combination with lenalidomide and dexamethasone and 317 patients to
lenalidomide plus dexamethasone. The most common adverse reactions
(greater than or equal to 20%), with an increased rate in the elotuzumab
arm compared to the control arm, were fatigue, diarrhea, pyrexia,
constipation, cough, peripheral neuropathy, nasopharyngitis, upper
respiratory tract infection, decreased appetite, and pneumonia.
Other important adverse reactions include infusion reactions,
infections, second primary malignancies, hepatotoxicity, and
interference with determination of complete response. As elotuzumab is
an IgG kappa monoclonal antibody, it can be detected in the serum
protein electrophoresis and immunofixation assays used to assess
response.
Serious adverse events occurred in 65.4% of patients in the
elotuzumab-containing arm compared to 56.5% in the lenalidomide plus
dexamethasone alone arm. The most common serious adverse reactions were
pneumonia, pyrexia, respiratory tract infection, anemia, pulmonary
embolism, and acute renal failure.
The recommended dose and schedule for elotuzumab is 10 mg/kg
intravenously every week for the first two cycles and every 2 weeks,
thereafter, until disease progression or unacceptable toxicity with
lenalidomide 25 mg daily orally on days 1 through 21. Dexamethasone is
administered as follows: In weeks with elotuzumab infusion,
dexamethasone is to be administered in divided doses, 8 mg intravenously
prior to infusion and 28 mg orally; in weeks without elotuzumab
infusion, dexamethasone is to be administered 40 mg orally.
Pre-medication with an H1 blocker, H2 blocker, and acetaminophen should
be administered prior to elotuzumab infusion.
Elotuzumab is being approved prior to the Prescription Drug User
Fee Act (PDUFA) goal date of February 29, 2016. This application was
granted priority review and had breakthrough therapy designation. A
description of these expedited programs is in the Guidance for Industry:
Expedited Programs for Serious Conditions-Drugs and Biologics,
available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358301.pdf
Full prescribing information is available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/761035s000lbl.pdf
Empliciti’s Cost
Empliciti will be sold in the U.S. in two vials sizes: A smaller vial that contains 300 mg of the drug, and a larger vial that contains 400 mg.
Bristol-Myers Squibb has informed The Beacon that the wholesale price per vial of Empliciti will be $1,776 for the 300 mg vial and $2,368 for the 400 mg vial.
Using these prices and an assumed patient weight of between 154 and 176 pounds, Empliciti will cost $18,944 per four-week cycle for each of the first two cycles of treatment, and $9,472 per cycle thereafter. This means, in turn, that Empliciti’s cost per year will be $142,080 in the first year and $123,136 in subsequent years.
In comparison, Velcade costs between $4,800 and $8,500 per four-week cycle, depending on how often it is dosed. Ninlaro costs $8,670 per four-week cycle. And Kyprolis costs $10,500 per four-week cycle at the standard (20 – 27 mg/m2) dose.
Additional details about the FDA approval of Empliciti can be found in this press release from the FDA, a related press release from Bristol-Myers Squibb and AbbVie, and the full Empliciti prescribing information.
The results of the ELOQUENT-2 trial were published in Lonial, S. et al., “Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma,” The New England Journal of Medicine, June 2, 2015 (abstract). Slides from the ASCO presentation summarizing the ELOQUENT-2 results can be viewed here (PDF, courtesy of Dr. Lonial). This Beacon news article provides an in-depth look at the trial results.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | SLAMF7 (CD319) |
| Clinical data | |
| Trade names | Empliciti |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
IV |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Identifiers | |
| CAS Number | 915296-00-3 |
| ATC code | None |
| IUPHAR/BPS | 8361 |
| UNII | 1351PE5UGS |
| Chemical data | |
| Formula | C6476H9982N1714O2016S42 |
| Molecular mass | 145.5 kDa |
References
1 “Press Announcement—FDA approves Empliciti, a new immune-stimulating therapy to treat multiple myeloma”. U.S. Food and Drug Administration. Retrieved 3 December 2015.
2“Empliciti (elotuzumab) for Injection, for Intravenous Use. Full Prescribing Information” (PDF). Empliciti (elotuzumab) for US Healthcare Professionals. Bristol-Myers Squibb Company, Princeton, NJ 08543 USA.
3 “Bristol-Myers Squibb and AbbVie Receive U.S. FDA Breakthrough Therapy Designation for Elotuzumab, an Investigational Humanized Monoclonal Antibody for Multiple Myeloma” (Press release). Princeton, NJ & North Chicago, IL: Bristol-Myers Squibb. 2014-05-19. Retrieved 2015-02-05.
///////




























{kind=link}