MOXIFLOXACIN
Bay-12-8039
186826-86-8 HYDROCHLORIDE
| 1-cyclopropyl-7-[(1S,6S)-2,8-diazabicyclo[4.3.0]non-8-yl]-6-fluoro-8-methoxy-4-oxo- quinoline-3-carboxylic acid |
(4aS-Cis) -l-cyclopropyl-7- (2, 8- diazabicyclo [4.3.0] non-8-yl) -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylic acid
ALSO AS….
Vigamox, Avelox I.V., 151096-09-2, Moxifloxacin [INN:BAN], MXFX, CHEMBL32, Actira (*Hydrochloride*), Avelox (*Hydrochloride*)
Molecular Formula: C21H24FN3O4
Molecular Weight: 401.431363
Moxifloxacin is a synthetic fluoroquinolone antibiotic agent. Bayer AG developed the drug (initially called BAY 12-8039) and it is marketed worldwide (as the hydrochloride) under the brand name Avelox (in some countries also Avalox) for oral treatment
For the treatment of sinus and lung infections such as sinusitis, pneumonia, and secondary infections in chronic bronchitis. Also for the treatment of bacterial conjunctivitis (pinkeye).
Moxifloxacin and its salts are antibacterial agents, which were disclosed in EP 550,903. Moxifloxacin, chemically 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS, 7aS)-octahydro-6H-pyrrolo[3,4-b]pyridine-6-yl]-4-oxo-3-quinolinecarboxylic acid
Moxifloxacin is a fourth-generation synthetic
fluoroquinolone antibacterial agent developed by Bayer AG (initially called BAY 12-8039). It is marketed worldwide (as the hydrochloride) under the brand names
Avelox,
Avalox, and
Avelon for oral treatment. In most countries, the drug is also available in parenteral form for intravenous infusion. Moxifloxacin is also sold in an ophthalmic solution (eye drops) under the brand names
Vigamox,
Moxezafor the treatment of conjunctivitis (pink eye).
A United States patent application was submitted on 30 June 1989, for Avelox (moxifloxacin hydrochloride).
[1] In 1999 Avelox was approved by the
U.S. Food and Drug Administration (FDA) for use in the United States.
[2]
In the United States, moxifloxacin is licensed for the treatment of acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, community acquired pneumonia, complicated and uncomplicated skin and skin structure infections, and complicated intra-abdominal infections.
[3]
In the European Union, it is licensed for acute bacterial exacerbations of chronic bronchitis, non-severe community-acquired pneumonia, and acute bacterial sinusitis. Based on its investigation into reports of rare but severe cases of liver toxicity and skin reactions, the European Medicines Agency recommended in 2008 that the use of the oral (but not the IV) form of moxifloxacin be restricted to infections in which other antibacterial agents cannot be used or have failed.
[4] In the US, the marketing approval does not contain these restrictions, though the label contains prominent warnings against skin reactions.
MOXIFLOXACIN
Avelox (moxifloxacin) was launched in the United States in 1999 and is currently marketed in more than 80 countries worldwide. In the United States, Avelox is marketed by Bayer’s partner Merck.
In 2011 the FDA added two boxed warnings for this drug in reference to spontaneous tendon ruptures and the fact that moxifloxacin may cause worsening of
myasthenia gravis symptoms, including muscle weakness and life-threatening breathing problems.
[5]
The initial approval by the FDA (December 1999)
[7] encompassed the following indications:
- Acute Exacerbations of Chronic Bronchitis (AECB)
- Acute Bacterial Sinusitis (ABS)
- Community Acquired Pneumonia (CAP)
Additional indications were approved by the FDA as follows:
- April 2001: Uncomplicated Skin and Skin Structure Infections (uSSSI)[8]
- May 2004: Community Acquired Pneumonia caused by multi-drug resistant Streptococcus pneumoniae.[9]
- June 2005: Complicated Skin and Skin Structure Infections (cSSSI)[10]
- November 2005: Complicated Intra-Abdominal Infections (cIAI).[11]
The European Union requires that moxifloxacin only be prescribed when other antibiotics that have been initially recommended for treatment cannot be used or have failed.
[12][13]
At the current time,
[when?] there are no approved uses within the pediatric population for Oral and I.V. moxifloxacin. A significant number of drugs found within this class, including moxifloxacin, are not licensed by the FDA for use in children due to the risk of permanent injury to the musculoskeletal system.
[14][15][16]
Note: Moxifloxacin may be licensed for other uses, or restricted, by the various regulatory agencies worldwide
Marketing authorisations for the tablet and injectable forms of Moxifloxacin are held by Bayer, while Alcon (now a subsidiary of Novartis) produces ophthalmic solutions for treating conjunctivitis under the brand names of Moxeza, Vigamox, and Moxivig. Avelox generated sales of USD320 million in the first 9 months of 2013.
Moxifloxacin is available in three distinct administration forms. Formulated as a salt, Moxifloxacin hydrochloride is sold as an oral 400 mg film-coated tablet and as an injectable solution for infusion by Bayer; although in the US it is distributed by Merck Sharp and Dohme under license from Bayer.
Alcon has formulated Moxifloxacin hydrochloride as a 0.5% ophthalmic solution under license from Bayer. Moxifloxacin was first discovered in 1988 and received the first market authorisation eleven years later in 1999 in the US.
Moxifloxacin hydrochloride is a synthetic broad-spectrum antibacterial agent. The active moiety, moxifloxacin has been shown to be clinically active against most strains of microorganisms such as aerobic gram-positive microorganisms including staphylococcus aureus, streptococcus pneumonia (penicillin-susceptible strains) and streptococcus pyogenes, aerobic gram-negative microorganisms including haemophilus influenza hemophilus parainfluenzae, klebisiella pneumonia. Moxifloxacin is commercially available under the brand name of AVELOX® marketed by Bayer pharms.
VIGAMOX® (moxifloxacin hydrochloride ophthalmic solution) 0.5% is a sterile solution for topical ophthalmic use. Moxifloxacin hydrochloride is an 8-methoxy fluoroquinolone anti-infective, with a diazabicyclononyl ring at the C7 position.
![VIGAMOX®<br /><br /><br /><br /><br /><br /><br /><br /><br /><br /><br /><br /><br /><br />
(moxifloxacin hydrochloride) Structural Formula Illustration” src=”<a href=]() http://images.rxlist.com/images/rxlist/vigamox1.gif” width=”286″ height=”171″ /> http://images.rxlist.com/images/rxlist/vigamox1.gif” width=”286″ height=”171″ /> |
C21H24FN304•HC1 Mol Wt 437.9
Chemical Name: l-Cyclopropyl-6-fluoro-l,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolol[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid, monohydrochloride. Moxifloxacin hydrochloride is a slightly yellow to yellow crystalline powder. Each mL of VIGAMOX® solution contains 5.45 mg moxifloxacin hydrochloride, equivalent to 5 mg moxifloxacin base.
Contains: Active: Moxifloxacin 0.5% (5 mg/mL); Inactives: Boric acid, sodium chloride, and purified water. May also contain hydrochloric acid/sodium hydroxide to adjust pH to approximately 6.8.
VIGAMOX® solution is an isotonic solution with an osmolality of approximately 290 mOsm/kg.
Market Considerations
Amongst the US approvals, Dr. Reddy’s, Teva, Torrent, and Aurobindo have received tentative approvals for the 400 mg oral tablet formulation. Akorn, Teva and Apotex have received tentative approvals for a Moxifloxacin hydrochloride ophthalmic solution. No 180 day period of exclusivity has been awarded since all patents were found to be valid.
In the UK, Teva, Rivopharm, and Double-E Pharma have received marketing authorisations for the 400 mg Moxifloxacin tablets, while Noridem has received a market authorisation for the equivalent 400mg/250ml solution for infusion. A similar trend of generic competition, for tablets and infusions, following molecule patent expiry is expected throughout Europe. Currently no generic market authorisations for ophthalmic formulations have been granted in major European countries. However, Sandoz and Hexal have gained market authorisations in some European markets for the ophthalmic dosage form following Novartis’ acquisition of Alcon.
In Canada the only generic manufacturer holding a marketing authorisation is Sandoz, however, this was granted as a New Drug Submission rather than as an ANDS. Following patent expiries from mid-2014, the European and North American markets are likely to have significant competition if the numbers of companies filing litigation, ANDS, ANDAs and the like in the northern hemisphere is anything to go by.

MOXIFLOXACIN
History
Moxifloxacin was first patented (United States patent) in 1991 by Bayer A.G., and again in 1997.
[47] Avelox was subsequently approved by the U.S. Food and Drug Administration (FDA) for use in the United States in 1999 to treat specific bacterial infections.
[2] Ranking 140th within the top 200 prescribed drugs in the United States for 2007
[48] moxifloxacin, in the same manner as
ciprofloxacin, has proven to be a blockbuster drug for Bayer A. G., generating billions of dollars in additional revenue. In 2007 alone, Avelox generated sales of $697.3 million dollars worldwide.
[26]
Patent
A United States patent application was made on 30 June 1989, for Avelox (moxifloxacin hydrochloride),(Bayer A.G. being the assignee), which was subsequently approved on 5 February 1991. This patent was scheduled to expire on 30 June 2009. However, this patent was extended for an additional two and one half years on 16 September 2004, and as such is not expected to expire until 2012.
[49] Moxifloxacin was subsequently (ten years later) approved by the
U.S. Food and Drug Administration (FDA) for use in the United States in 1999. There have been at least four additional United States patents filed regarding moxifloxacin hydrochloride since the 1989 United States application,
[47][50] as well as patents outside of the USA.
Additional regulatory history
6/12/2002 Changes made to minimize the impact of warnings concerning adverse reactions.
[51]
26 June 2003 New Zealand Pharmacovigilance warns of moxifloxacin induced respiratory insufficiency.
[52]
10/6/2003 Changes made to minimize the impact of post marketing reports as well as the risk of tendon injuries.
[53]
29 December 2008 Addition of numerous adverse reactions associated with the use of moxifloxacin.
[54]
27 April 2009 Issuance of a Medication Guide and revisions to include new safety information including the addition of the Black Box Warning to the Medication Guide. The FDA had determined that Moxifloxacin poses a serious and significant public health concern, requiring the distribution of a Medication Guide.
[55]
24 June 2009 Updating of the carton and container labels to include a statement to let dispensers know that a Medication Guide
must be dispensed with the product.(emphasis added)
[56]
Patent related
| COUNTRY | PATENT NUMBER | APPROVED | EXPIRES (ESTIMATED) |
|---|
| Canada | 1340114 | 1998-11-03 | 2015-11-03 |
| Canada | 2342211 | 2009-05-26 | 2019-09-29 |
| United States | 4990517 | 1994-12-08 | 2011-12-08 |
| United States | 6548079 | 2000-07-25 | 2020-07-25 |
|
As indicated by the Key Patent Indicator (Fig. 2), patents in the families with priority DE3824072A, 15/07/1988 (‘072), and DE4200414A, 10/01/1992, (‘414) provide protection for the Moxifloxacin molecule and are considered to be the main constraint to generic entry. As patents in the ‘414 family have expired or were never granted, the only remaining constraint to generic entry is the ‘072 family. The term of the Australian patent of this family have been extended to 19 June 2014 while the Canadian member will enjoy the longer term of 17 years from grant, expiring in November 2015.
Supplementary protection certificates (SPCs) have been granted in France, Germany, Spain and the UK, and will expire in June 2014. Given that there are less than 2 years until these SPCs expire, and that as yet no applications for paediatric extension of the SPCs have been published in Europe, they are unlikely to be extended by 6 months on the basis of the approved Paediatric Investigation Plans.
The US member, 4,990,517 (‘517), protecting the general structure of the Moxifloxacin molecule, expired in June 2012, after enjoying 6 month paediatric extension on top of a 901 day s156 extension. However, the absence of generics on the US market is due to Bayer securing a divisional patent, 5,607,942 (‘942). This patent claims the Moxifloxacin molecule specifically and is due to expire in September 2014, after being awarded a 6 month paediatric extension.
Members of the ‘072 family from both Canada and the US have been the subject of litigation after generic manufacturers identified these patents in paragraph IV filings, and the equivalent in Canada, as early as 2006. After filing an infringement suit in the US against Teva in relation to US ‘517, US ‘942, Bayer enjoyed a satisfying validification of their patents when Teva agreed that it would be infringing two of the patents, while the third was decided by the court to be equally valid.
In Canada, Novopharm, Cobalt, Apotex, Mylan and Apotex have also all tested the litigation waters relating to the equivalent patent with no success noted so far.
A third patent family that promises to be a constraint for generic ophthalmic formulations is Alcon’s 1998 patent, US10250498P (Fig. 2), which identifies an ophthalmic formulation of Moxifloxacin and its use in the treatment and prevention of eye infections. Patents in this family are set to expire in August 2019. US members 6,716,830 (‘830) and 7,671,070 (‘070) have been awarded a 6 month paediatric extension, extending their expiration until March 2020. The validity of US ‘830 was upheld following Teva filing paragraph IV certifications to manufacture generic Vigamox. Teva has since appealed this ruling.
In addition, Alcon has filed patent infringement suits against Watson, Lupin and Apotex in relation to US ‘070 after they submitted Abbreviated New Drug Applications (ANDAs)with paragraph IV filings in preparation for commercialisation of a Moxeza/Vigamox generic equivalent. There has been no outcome from these suits to date. In addition, applications for Orders of Prohibition against Cobalt, Apotex and Teva have also been noted for the equivalent Canadian patent following the filing of Abbreviated New Drug Submissions (ANDS) by these companies.
The equivalent European patent 1,117,401 was revoked following opposition by Teva filed in the European patent office. Its divisional patents, 1,384,478 (granted) and 2,301,541 (accepted) have restricted claims to the use of Moxifloxacin in the topical treatment of ophthalmic infections caused by P. aeruginosa and H. influenza, respectively. This may provide a prepared generic competitor an opportunity to launch their Moxifloxacin ophthalmic equivalent in Europe soon after the expiry of patents protecting the molecule, subject to legal review of the remaining claims of the patent.
Alcon have secured additional protection for their Moxeza ophthalmic formulation by way of patents in the family with priority US5987708P (09/06/2008). Patent claims specify ratios of Moxifloxacin to inactive ingredients and additional inactive ingredients and therefore generic competitors are likely to circumvent the patent by reformulation. Lupin has filed paragraph IV certifications to US8450311, which is currently subject of a patent infringement suit.
Families with priorities DE19546249A (12/12/1995), DE19751948A (24/11/1997), DE19855758A (10/11/1998) and US36433499A (30/07/1999) are not considered to be a constraint to generic entry because the protected technologies are likely to be circumvented.
Generic equivalents
In 2007, the U.S. District Court for the District of Delaware held that two Bayer patents on Avelox (moxifloxacin hydrochloride) are valid and enforceable, and infringed by Dr. Reddy’s ANDA for a generic version of Avelox.
[70][71] The district court sided with Bayer, citing the Federal Circuit’s prior decision in Takeda v. Alphapharm
[72] as “affirming the district court’s finding that defendant failed to prove a prima facie case of obviousness where the prior art disclosed a broad selection of compounds, any one of which could have been selected as a lead compound for further investigation, and defendant did not prove that the prior art would have led to the selection of the particular compound singled out by defendant.” According to Bayer’s press release
[70] announcing the court’s decision, it was noted that Teva had also challenged the validity of the same Bayer patents at issue in the Dr. Reddy’s case. Within Bayer’s first quarter 2008 stockholder’s newsletter
[73]
Bayer stated that they had reached an agreement with Teva Pharmaceuticals USA, Inc., the adverse party, to settle their patent litigation with regard to the two Bayer patents. Under the settlement terms agreed upon, Teva would obtain a license to sell its generic moxifloxacin tablet product in the U.S. shortly before the second of the two Bayer patents expires in March 2014.
- Economic impact: adverse reactions:
The advocacy group Public Citizen has lobbied for increasing safety warnings and for the removal of some fluoroquinolone drugs from clinical practice.
[74][75][76][77][78][79][80][81]
|
|
3-5-1997
|
7-(1-pyrrolidinyl)-3-quinolone- and – naphthyridone-carboxylic acid derivatives as antibacterial agents and feed additives
|
|
|
6-9-2000
|
INHIBITORS OF MULTIDRUG TRANSPORTERS INHIBITORS OF MULTIDRUG TRANSPORTERS
|
|
|
5-19-2000
|
PHARMACEUTICAL MOXIFLOXACIN PREPARATION
|
|
|
5-12-2000
|
AQUEOUS DRUG FORMULATION FOR ORAL APPLICATION AQUEOUS DRUG FORMULATION FOR ORAL APPLICATION
|
|
|
4-7-2000
|
ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE
|
|
|
4-7-2000
|
ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE
|
|
|
4-7-2000
|
ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE
|
|
|
1-20-2000
|
COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS
|
|
|
12-17-1999
|
NOVEL THERAPEUTIC AGENTS THAT MODULATE ENZYMATIC PROCESSES
|
|
|
4-2-1999
|
MEDICAMENT FORMULATION WITH A CONTROLLED RELEASE OF AN ACTIVE AGENT
|
|
|
8-28-1998
|
COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS
|
| | |
|---|
|
|
11-10-2006
|
Amorphous moxifloxacin hydrochloride
|
|
|
10-4-2006
|
Method for producing 8-methoxy-quinolinecarboxylic acids
|
|
|
7-15-2005
|
Pharmaceutical composition
|
|
|
7-13-2005
|
Aqueous pharmaceutical composition containing moxifloxacin or salts thereof
|
|
|
5-25-2005
|
Method for producing 8-methoxy-quinolinecarboxylic acids
|
|
|
2-6-2004
|
Medicinal composition
|
|
|
5-21-2003
|
Method for the enantiomer separation of cis-8-benzyl-7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane
|
|
|
11-31-2000
|
OPTICALLY ACTIVE QUINOLINE CARBOXYLIC ACID DERIVATIVES HAVING 7-PYRROLIDINE SUBSTITUTES CAUSING OPTICAL ACTIVITY AND A PROCESS FOR PREPARING THEREOF
|
|
|
11-17-2000
|
ANTIBACTERIAL OPTICALLY PURE BENZOQUINOLIZINE CARBOXYLIC ACIDS, PROCESSES, COMPOSITIONS AND METHODS OF TREATMENT (S)-BENZOQUINOLIZINE CARBOXYLIC ACIDS AND THEIR USE AS ANTIBACTERIAL AGENTS
|
|
|
8-32-2000
|
COMPOSITIONS AND METHODS FOR IMPROVED DELIVERY OF HYDROPHOBIC THERAPEUTIC AGENTS
|
| | |
|---|
|
|
6-18-2010
|
MULTI-ARM POLYMER PRODRUGS
|
|
|
4-9-2010
|
COMPOSITION COMPRISING AN ANTIBIOTIC AND A CORTICOSTEROID
|
|
|
7-22-2009
|
Tri-, tetra-substituted-3-aminopyrrolidine derivative
|
|
|
7-3-2009
|
NOVEL HYDRATE FORM
|
|
|
3-20-2009
|
Multi-Arm Polymer Prodrugs
|
|
|
1-16-2009
|
Sulfonamide Derivatives for the Treatment of Bacterial Infections
|
|
|
11-21-2008
|
Phosphonated Fluoroquinolones, Antibacterial Analogs Thereof, and Methods for the Prevention and Treatment of Bone and Joint Infections
|
|
|
8-15-2008
|
Multi-arm polymer prodrugs
|
|
|
8-24-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-29-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
7-4-2012
|
TRI-, TETRA-SUBSTITUTED-3-AMINOPYRROLIDINE DERIVATIVE
|
|
|
6-13-2012
|
Process for the Synthesis of Moxifloxacin Hydrochloride
|
|
|
12-2-2011
|
COMPACTED MOXIFLOXACIN
|
|
|
10-12-2011
|
Treatment of bacterial diseases of the respiratory organs
|
|
|
9-16-2011
|
Novel Hydrate Form
|
|
|
9-2-2011
|
NOVEL POLYMORPH OF MOXIFLOXACIN HYDROCHLORIDE
|
|
|
6-31-2011
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-10-2011
|
SYNTHESIS OF (4aS,7aS)-OCTAHYDRO-1H-PYRROLO[3,4-b]PYRIDINE
|
|
|
7-30-2010
|
MULTI-ARM POLYMER PRODRUGS
|
|
|
6-30-2010
|
Multi-arm polymer prodrugs
|
DESCRIPTION
Moxifloxacin is a therapeutic agent that shows a broad spectrum antibacterial action. Moxifloxacin is the international non-proprietary name (INN) for 1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid.
The structure of moxifloxacin corresponds to formula (I):
Racemic moxifloxacin was firstly described in EP-A-350733 and, particularly, moxifloxacin having a (S,S)-configuration is described inEP-A-550903 .
In experimental example 19 of EP-A-550903 and example Z19 of EP-A-591808 , a method for preparing and isolating moxifloxacin base is described. The same method is described in patent document EP-A-592868 ). These published patent applications neither describe nor suggest the possible existence of a crystalline form of moxifloxacin base. In
WO-A-2008059521 it is disclosed that by performing the mentioned examples an acetonitrile solvated form of moxifloxacin with low purity is obtained, and so the obtained solvate form can not be used as such in pharmaceutical formulations. In the above mentioned examples, the obtained moxifloxacin base crude is purified and isolated by chromatography using methylene chloride/methanol/17% aqueous ammonia as the solvent system. The purification process disclosed in said documents has been reproduced by the authors of the present invention, but only an amorphous form of moxifloxacin was obtained. This process for the purification and isolation of moxifloxacin base is complex and difficult to perform on industrial scale due to the need for purifying the product by column chromatography.
WO-A-9926940 discloses a process for the preparation of moxifloxacin from a difluoro precursor comprising the step of adjusting the pH to 6.8- 7.0. However, the reproduction of this example shows that at this pH moxifloxacin hydrochloride or a mixture of moxifloxacin hydrochloride and moxifloxacin base is obtained, since the X-ray diffractogram of the isolated compound corresponds with the hydrochloride moxifloxacin. This fact has also been confirmed by the reproduction of the subsequent crystallization described in the international patent application of the previous compound in ethanol/water. The behaviour of this solid was very different regarding its solubility in respect what was described in the international patent application.
WO-A-2004091619 and
WO-A-2007010555 disclose the preparation of moxifloxacin base as a solid. Nevertheless, they do not describe nor suggest the preparation of a crystalline form of moxifloxacin base. The authors of the present invention have proved that the X-ray diffractogram of the product obtained by reproducing the reference example disclosed in
WO-A-2004091619 , based on a pH adjustment to 7.0-7.2, corresponds to the X-ray diffractogram of the monohydrate of moxifloxacin hydrochloride disclosed in
US 5849752 . Similarly, by reproducing the reference example of
WO-A-2007010555 , also for obtaining moxifloxacin base but based on a pH adjustment to 5.0-6.0, it is neither expected to obtain moxifloxacin base due to the adjustment of the solution within a range of acidic pH values.
[0008]
WO-A-2008059521 discloses a process for the preparation of a crystalline form of moxifloxacin base, designated as Form I, by recrystallization in a ketosolvent, such as acetone. This form is characterized by its X-ray diffraction pattern corresponding to a hemihydrate. The best yield obtained is a 78.2% starting from moxifloxacin hydrochloride in example 18. This crystalline form has a tendency to occlude solvent molecules within the crystalline network in amounts very superiors to the allowed ones by for Guidelines Residual Solvents (CPMP/ICH/283/95) and can be difficult to impossible to remove by drying, what forces to carry out laborious treatments, either physical, or chemical to reach allowed solvent levels. The presence of any non-aqueous solvents in amounts over the allowed ones would not make this crystalline form suitable for the preparation of pharmaceutical formulations.
The existence of polymorphs is unpredictable and there is no a priori established procedure to prepare an unknown polymorph. The difference in the physical properties of different morphological forms results from the orientation and intermolecular interactions of adjacent molecules are complexes in the bulk solid. Furthermore, the different solid forms of a pharmaceutically active ingredient can have different characteristics, and offer certain advantages in methods of manufacture and also in pharmacology. Thus, the discovery of new solid forms can contribute to clear improvements in the efficiency of methods of production and/or improvements in the characteristics of the pharmaceutical formulations of the active ingredients, since some forms are more adequate for one type of formulation, and other forms for other different formulations.
Moxifloxacin Hydrochloride namely (4aS-Cis) -l-cyclopropyl-7- (2, 8- diazabicyclo [4.3.0] non-8-yl) -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylic acidhydrochloride has the formula
Moxifloxacin Hydrochloride
Moxifloxacin is a fluoroquinolone broad spectrum antibacterial particularly against Gram-positive bacteria significantly better than those of Sparfloxacin and Ciprofloxacin that was disclosed in EP No 350,733 and EP No 550,903. Moxifloxacin has activity against Gram- negative and Gram-positive organisms, including Streptococcus pneumonia, Staphylococcus aureus, Pseudomonas aeruginosa, particularly against the respiratory disease-causing pathogens like Mycoplasma pneumonia, Mycobacterium tuberculosis, Chlamydia pneumoniae and the activity shown to be unaffected by B-lactamases .
US Patent No 5,157,117 discloses (l-cyclopropyl-6, 7-difluoro-8-methoxy- 4-oxo-l, 4-dihydro-3-quinoline carboxylic acid-O3, 04)bis (acyloxy-O) borate and process for its preparation by reacting ethyl-1- cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3-quinoline carboxylate with Boric acid and acetic anhydride in presence of zinc chloride and its conversion to Gatifloxacin hydrochloride.
WO 2005/012285 discloses the process for the preparation of moxifloxacin hydrochloride using a novel intermediate namely (4aS-Cis)-(1-cyclopropyl-7-(2,8-diazabicyclo[4,3,0]non-8-yl)-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-3-quinoline carboxylic acid-O3,O4)bis(acycloxy-O)borate.
Hydrates of Moxifloxacin hydrochloride known are the anhydrous and monohydrate. US Patent No. 5,849,752 discloses the monohydrate of Moxifloxacin hydrochloride and its preparation by treating the anhydrous crystalline form with ethanol/ water mixtures.
The prior art disclosed in European Patent No’s EP 350,733, EP 550,903 and EP 657,448 discloses the preparation of Moxifloxacin hydrochloride involving the condensation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid or its esters with (S,S) 2,8-Diaza bicyclo [4.3.0] nonane in presence of a base and its conversion to hydrochloride at higher temperatures leading to the desired Moxifloxacin along with its positional isomer namely (4aS-Cis)-l- cyclopropyl-6- (2, 8-diazabicyclo [4.3.0] non-8-yl) -7-fluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid as a major impurity. As the impurity and the Moxifloxacin are positional isomers they are difficult to separate. Purification of Moxifloxacin to remove this isomer results in lower yields thereby increasing the product cost. Similarly methods described in the prior art involves the preparation of Moxifloxacin and then its conversion to its hydrochloride thereby incorporating an additional step in the manufacturing process also leading to lowering of yields.
Moxifloxacin and its pharmacologically acceptable salts are disclosed in European patents EP 350733, EP 550903 and EP 657,448. The disclosed process for the preparation of moxifloxacin hydrochloride comprises of condensing l-cyclopropyl-6,7- difluoro-8-methoxy-4-oxo-l,4-dihydro-3-quinoline carboxylic acid or its esters with (S,S)2,8-diazobicyclo[4.3.0]nonane, in presence of a base at high temperature followed by conversion into hydrochloride salt .
This process not only produces desired moxifloxacin hydrochloride but also its positional isomer namely l-cyclopropyl-7-fluoro- 1,4- dihydro -8- methoxy -6- (4aS,7aS)- octahydro- 6H -pyrrolo [3,4-b] pyridine-6-yl] -4- oxo-quinolinecarboxylic acid as a major impurity which is difficult to separate. The purification of moxifloaxcin to remove this isomer results in lower yields thereby increasing the product cost.
The International publication WO 2005/012285 discloses an improved process for the preparation of moxifloxacin hydrochloride incorporated herein by reference. The disclosed process involves the preparation of moxifloxacin hydrochloride from the ethyl 1 -cyclopropyl-6,7-difluoro-8-methoxy-4-oxo- 1 ,4-dihydro-3-quinolme carboxylate through a novel intermediate (4aS-cis)-l-cyclopropyl-7-(2,8-diazabicyclo[4.3.0]non-8- yl)-6-fluoro-8-methoxy-4-oxo- 1 ,4-dihydro-3 -quinolinecarboxylicacid-03,04)bis(acyloxy -0)-borate.
US patent application 6897315 discloses a process for the preparation of 8-methoxy-3-quinoline carboxylic acid especially moxifloxacin incorporated herein by reference. The disclosed process involves the preparation of moxifloxacin from 8-halo moxifloxacin derivative using methanol and potassium tertiary butoxide. US patent 5639886 discloses one-pot process for the preparation of 3-quinoline carboxylic acid derivatives including moxifloxacin.
WO 2004 091619 claims anhydrous crystalline form-Ill of moxifloxacin hydrochloride and WO 2004/039804 claims amorphous form of moxifloxacin hydrochloride. US Pat.No.5, 849,752 discloses specific crystalline forms of anhydrous moxifloxacin mono hydrochloride and monohydrated moxifloxacin mono hydrochloride. Anhydrous moxifloxacin mono hydrochloride disclosed in US Pat. No.5, 849,752 has been designated as “Form-I” and the hydrated form as “Form-II” in US Pat. No.7,230,006. It also discloses a novel crystalline Form-Ill of anhydrous moxifloxacin mono hydrochloride.
US patent US 5,480,879 discloses the melting range of moxifloxacin in example part as 203-208°C and does not speaks about polymorphism of moxifloxacin. Experiment executed as per the procedure given in example Zl 9 of US 5,480,879 and resulted in acetonitrile solvated form of moxifloxacin with low purity and the obtained solvated form can not used for formulations
U.S. Pat. No. 5,849, 752 (“the ‘752 patent”), incorporated by reference, described two crystalline forms of moxifloxacin hydrochloride namely, anhydrous moxifloxacin hydrochloride and monohydrated moxifloxacin hydrochloride. For convenience, the anhydrous crystalline form described in the 752 patent is designated as “Form I”, and the hydrated form as “Form II”. According to U.S. Pat. No. ‘752’, moxifloxacin hydrochloride monohydrate Form II was obtained by stirring a suspension of the anhydrous moxifloxacin hydrochloride in aqueous media until hydration. Moxifloxacin hydrochloride monohydrate of ‘752’ was also prepared by crystallizing moxifloxacin hydrochloride from a media having a water content which is stoichiometrically sufficient but limited to 10%.
WO patent application publication No. 04/091619 disclosed anhydrous Form III of moxifloxacin hydrochloride.
WO patent application publication No. 04/039804 disclosed amorphous form of moxifloxacin hydrochloride.
WO 2005/054240 disclosed two novel crystalline forms which were designated as Form A and Form B of moxifloxacin hydrochloride.
WO patent application publication No. 07/010555 disclosed two crystalline forms which were Form X and Form Y of moxifloxacin hydrochloride. According to WO Publication No. 2007/010555, Form Y was obtained by crystallization of moxifloxacin hydrochloride from the mixture of methanol and water in the ratio of about 8:1 by volume.
WO patent application publication No. 07/148137 disclosed hydrate form of moxifloxacin hydrochloride. According to WO Publication No. 2007/148137, moxifloxacin hydrochloride monohydrate was obtained by crystallization moxifloxacin hydrochloride by humidification of moxifloxacin hydrochloride at 50-90% relative humidity at 25-60° C. for 8 to 24 hours.
WO patent application publication No. 08/028959 disclosed crystalline form of moxifloxacin hydrochloride. According to WO Publication No. 2008/028959, moxifloxacin hydrochloride was obtained by dissolving moxifloxacin hydrochloride in a mixture of methanol and water and adding acetone and recovering moxifloxacin hydrochloride crystalline form.
WO patent application publication No. 08/059521 disclosed process for the preparation of anhydrous crystalline form I of moxifloxacin hydrochloride.
WO patent application publication No. 08/095964 disclosed crystalline form of moxifloxacin base.
……………………
SYNTHESIS
Drugs Fut 1997,22(2),109
36th Intersci Conf Antimicrob Agents Chemother (Sept 15-18, New Orleans) 1996,Abst. F1.
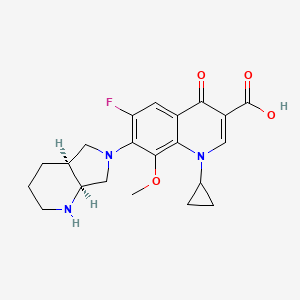
The anhydrization of pyridine-2,3-dicarboxylic acid (I) with acetic anhydride gives the corresponding anhydride (II), which by treatment with benzylamine (III) is converted into the benzylimide (IV). The hydrogenation of (IV) with H2 over Pd/C yields 8-benzyl-2,8-diazabicyclo[4.3.0]nonane-7,9-dione (V), which is further hydrogenated with LiAlH4, affording (?-cis-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VI) (1). The optical resolution of (VI) by separation of the cis-(R,R)-isomer as crystalline L-(+)-tartrate and further purification of the cis-(S,S)-isomer (VII) as the D-(-)-tartrate affords enantiomerically pure (S,S)-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VII). The debenzylation of (VII) by hydrogenolysis with H2 over Pd/C gives (S,S)-2,8-diazabicyclo[4.3.0]nonane (VIII), which is condensed with 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) in basic medium and finally salified with HCl. The 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) has been obtained as follows: The reaction of 2,4,5-trifluoro-3-methoxybenzoyl chloride (X) with malonic acid monoethyl ester monopotassium salt (XI) by means of triethylamine gives 2-(2,4,5-trifluoro-3-methoxybenzoyl)acetic acid ethyl ester (XII), which is condensed with triethyl orthoformate yielding the corresponding ethoxymethylene derivative (XIII). The reaction of (XIII) with cyclopropylamine affords the cyclopropylaminomethylene derivative (XIV), which is finally cyclized to (IX) by means of NaF in DMF.
………………………..
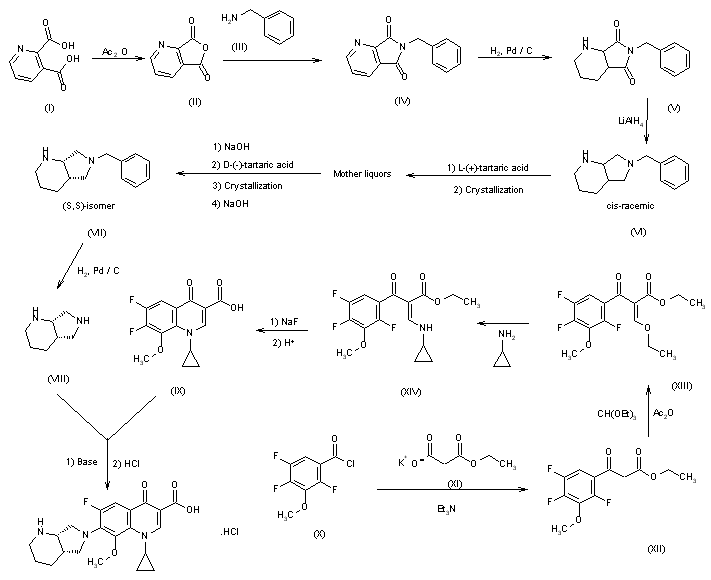
J Label Compd Radiopharm 2000,43(8),795
The condensation of 2,4,5-trifluoro-3-methoxybenzoyl chloride (I) with 14C-labeled diethyl malonate (II) by means of MgCl2 and TEA gives the benzoylmalonate (III), which is monodecarboxylated with TsOH in refluxing water, yielding the benzoylacetate (IV). The reaction of (IV) with triethyl orthoformate and Ac2O at 140 C affords the benzoylacrylate (V), which is treated with cyclopropylamine (VI) in cyclohexane to provide ethyl 3-(cyclopropylamino)-2-(2,4,5-trifluoro-3-methoxybenzoyl)acrylate (VII). The cyclization of (VII) by means of K2CO3 in hot N-methylpyrrolidone gives the quinolone carboxylate (VIII), which is hydrolyzed with NaOH in hot methanol, affording the carboxylic acid (IX). Finally, this compound is condensed with (S,S)-2,8-diazabicyclo[4.3.0]octane (X) by means of 1,4-diazabicyclo[2.2.2]octane (DABCO) in refluxing acetonitrile.
…………………….
……………………
The reaction scheme is given below: Stage-I
Acetic anhydride
Ethyl-l-cyclopropyl-6, 7- ( l-cyclopropyl-6, 7-difluoro-l, 4- difluoro-1, 4-dihydro-8- dihydro-8- methoxy-4-oxo-3- methoxy-4-oxo-3-quinoline quinoline carboxylic acid-03, 04) carboxylate . Bis ( acetate-O) -borate (Borate complex)
Stage-II
Triet yl amine
Acetonitrile
l-cyclo propyl-6, 7-difluoro- [S, S] -2, 8-diazabicyclo- (1- cyclo propyl-6, fluoro-7 (2, 8- 1, 4-dihydro-8- methoxy-4-oxo [4 ,3.0]nonane Diazabicyclo-nonane) 1,4- -3-quinoline carboxylic acid- dihydro-8-methoxy-4-oxo-3 03,04)Bis ( acetate-O) -borate- quinoline carboxylic acid- (Borate complex) (03,04) bis (acetate-O) -borate itage-III
(1- cyclo propyl-6, fluoro- (2, 8- Moxifloxacin HCI pseudohydrate Diazabicyclo-nonane) 1,4- dihydro-8-methoxy-4-oxo-3 -quinolinecarboxylicacid- (O^O4) bis (acetate-O) -borate
Stage-TV
Moxifloxacin HCI pseudohydrate Moxifloxacin HCI monohydrate
EXAMPLE – I
Stage-1: Preparation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo- l,4-dihydro-3-quinoline carboxylic acid-O3,O*)bis (acyloxy-O)borate
Acetic anhydride (175 g) is heated to 70°C and boric acid (30 g) is slowly added lot wise in a temperature range of 70°C to 90°C. The temperature is then raised, maintained under reflux for 1 hr followed by cooling to about 70°C. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylate (100 g) is added under stirring. The temperature is then raised and maintained for 1 hr in the range of 100°C to 105°C. The reaction mass is cooled to 0°C, chilled water (400 ml) is added slowly followed by cold water (600 ml) at temperature 0°C to 5°C and maintained for 2 hrs at 0°C to 5°C. The product which is a boron acetate complex is filtered, washed with water (500 ml) and dried at 55°C to 60°C under vacuum to constant weight. The dry wt is 130.0 g corresponding to yield of 95.2%.
Stage-2: Preparation of (4aS-Cis) -l-Cyclopropyl-7- (2, 8-diazabicyclo [4.3.0]non-8-yl) -6-fluoro-8-methoxy-4-oxo-l , 4-dihydro-3-quinoline carboxylicacid-03,0*)bis (acyloxy-O)borate
The boron acetate complex (130 g) prepared in stage 1 is suspended in acetonitrile (650 ml), and [S, S] -2, 8-diazabicyclo [4.3.0] nonane (47 g) and triethyl amine (72.9 g) are added. The temperature is raised to reflux and maintained for 1 hr. at reflux, followed by cooling to about 40°C. The solvent is removed under vacuum at temperature below 40°C, and n-hexane (200 ml) is added. After maintaining the reaction mass for 1 hr at room temperature the product is isolated by filtration followed by washing of the wet cake with n-hexane . The product is dried at about 45°C to about 50°C to constant weight.
Dry wt of the novel intermediate is 117.0 g corresponding to yield of 71.5%.
Elemental analysis: C: 56.42%, H: 5.62%, N: 7.76% and the calculated values for the intermediate, formula C25H29BFN308C: 56.6%, H: 5.47%, N: 7.92%
IR Spectrum (KBr, cm-1) : 3415, 3332, 2936, 1718, 1630, 1573, 1526, 1445, 1273, 1042, 935, 860, 798, 682
^ NMR (200 MHz, CDC13, ppm) : 9.00 (1H), 7.82 (1H), 4.12 (4H), 3.57 (3H), 3.43 (4H), 3.07 (2H) , 2.75 (2H), 2.4 (1H),’ 2.1 (6H), 1.84 (2H) , 1.6 (1H), 1.31 (2H)
Mass Spectrum (MJ : 530.3 [M+H] , 470.2 [M+ - CH3COOH] , 428.2 [M+- (CH3CO)20, 100%], 402.2, 388.2
Stage -3: Preparation of Moxifloxacin Hydrochloride pseudohydrate
The intermediate (117 g) prepared stage-2 is dissolved in ethanol (600 ml) by stirring for about 30 min. at room temperature and the insolubles if any are filtered off. pH of the filtrate is adjusted to about 0.5 by addition of hydrochloric acid at room temperature and maintained for 2 hrs. The reaction mass is cooled, and maintained for two hrs, at about 0°C to about 5°C. The product is filtered, washed with chilled ethanol (50 ml) and dried at about 50°C to about 55°C till constant weight.
The dry weight of the Moxifloxacin hydrochloride pseudohydrate is 87.5g corresponding to yield of 91.0%. Water content of the product by KF is 0.64% w/w.
X-ray diffraction pattern data are given in Table-1 EXAMPLE – II
Stage- 2 : Preparation of Moxifloxacin pseudohydrate with out isolating (4aS-Cis) -l-Cyclopropyl-7- (2 , 8-diazabicyclo [4.3.0] on-8-yl) -6-fluoro- 8-methoxy-4-oxo-l,4-dihydro-3-quinolinecarboxylicacid-03,04)bis (acyloxy-O) borate
The boron acetate complex (130 g) prepared in stage-1 of Example-1 is suspended in acetonitrile (650ml) and [S, S] -2, 8-Diazabicyclo [4.3.0]nonane (47 g) & triethyl amine (72.9 g) are added. Temperature of the reaction mass is raised to reflux, maintained for 1 hr. at reflux and cooled to room temperature. Methanol (600 ml) is added and maintained for 30 min at room temperature to obtain a clear solution. The solution is filtered to remove insolubles if any and pH of the filtrate is adjusted to about 0.5 with hydrochloric acid (57.5 g) . The reaction mass is maintained for 2 hrs at temperature in the range of about 20°C to about 25°C, cooled to 0°C followed by maintaining the reaction mass at about 0°C to about 5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at about 50°C to 55°C until constant weight.
Dry wt of the Moxifloxacin hydrochloride pseudohydrate is 88g corresponding to yield of 68.7%.
EXAMPLE – III : Preparation of Moxifloxacin Hydrochloride monohydrate
Moxifloxacin hydrochloride (50 g) prepared as above is suspended in a mixture of ethanol (250 ml) and hydrochloric acid (25 ml) . Raised the temperature, maintained for two hrs at 40°C to 45°C followed by cooling to about 25°C. The product is filtered and dried under vacuum at 50-55°C until become constant weight.
Dry wt of Moxifloxacin hydrochloride monohydrate is 46 g corresponding to yield of 90.5%.
The IR spectral data and XRD pattern are identical with available Moxifloxacin hydrochloride monohydrate.
……………………….
Synthesis
The preferred embodiments of the present invention is represented as follows
Formula-2a Formula-2b
Formula-3a Formula-3b Formula-3c Formula-3d
Formula-4b
Formula-4c
SCHEME-5:
SCHEME-6:

Example-1: Preparation of (S,S)-2-benzyl-8-trityI-2,8-diazabicyclo (4.3.0) nonane:
A mixture of (S5S)-2-benzyl-2,8-diazabicyclo-(4.3.0) nonane 50 grams, dichloromethane 300 ml and triethylamine 28 ml was stirred at 25-35°C. Trityl chloride 71 grams was added to the reaction mixture. The reaction mixture was stirred at 25-350C for 5 hrs. The organic layer was washed with 5% sodium bicarbonate solution followed by water. The organic layer was distilled off completely to provide the title compound as a residue.
Yield: 96 grams
Example-2:
Preparation of (S,S)-8-trityI-2,8-diazabicyclo (4.3.0) nonane:
A mixture of (S,S)-2-benzyl-8-trityl-2,8-diazabicyclo (4.3.0) nonane 130 grams,
2- butanol 1600 ml and 5% Pd/C was taken in an autoclave and heated to 45-50°C at 4.0-4.5 Kg/cm2 of hydrogen pressure. The reaction mixture was stirred at this condition for 45-50 hrs. The reaction mixture was cooled to 25-300C and filtered through hyflow bed. The solvent was distilled off completely to provide the title compound as a residue.
Yield: 95 grams.
Example-3:
Preparation of condensed compouad of formula-4a:
A mixture of 20 grams of l-cyclopropyl-NjN-diethyl-όjT-difluoro-l^-dihydro-δ- methoxy-4-oxoquinoline-3-carboxamide, 11.8 grams of (S,S)2,8-diazabicyclo (4.3.0) nonane, 100 ml of acetonitrile and 2 grams of l,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) was heated to 80
0C. The reaction mixture was stirred for 35 hours at
 0
0C. The reaction mixture was cooled to 32
0C and stirred for 45 minutes at 32°C. The solvent was distilled off under reduced pressure. Water (100 ml) was added to the obtained residue. The reaction mixture was cooled to 25-35
0C. The reaction mixture was extracted with ethyl acetate. The solvent was distilled off to get the title compound as a residue. Yield: 12 grams.
Example-4:
Preparation of condensed compound of formula-4b:
A mixture of 5 grams of l-cyclopropyl-NjN-diethyl-όJ-difluoro-M-dihydro-δ- methoxy-4-oxoquinoline-3-carboxamide, 2.9 grams of (S,S)-8-phenyloxy carbonyl-2,8- diazabicyclo (4.3.0) nonane, 25 ml of acetonitrile and 0.5 grams of diazabicyclo[5.4.0]undec-7-ene (DBU) was heated to 800C. The reaction mixture was stirred for 35 hours at 75-8O0C. The reaction mixture was cooled to 25-35°C. The reaction mixture was stirred for 45 minutes at 25-35°C. The solvent was distilled off under reduced pressure. Water (100 ml) was added to the obtained residue. The reaction mixture was cooled to 25-35°C. The reaction mixture was extracted with ethyl acetate. The solvent was distilled off to get the title compound as a residue. Yield: 1 gram.
Example-5: Preparation of condensed compound of formula-4c:
A mixture of 5 grams of l-cyclopropyl-NjN-diethyl-βJ-difluoro-l^-dihydro-S- methoxy-4-oxoquinoline-3-carboxamide, 2.9 grams of (S,S)-8-tertiary butyloxycarbonyl- 2,8-diazabicyclo (4.3.0) nonane, 25 ml of acetonitrile and 0.5 grams of diazabicyclo[5.4.0]undec-7-ene (DBU) was heated to 800C. The reaction mixture was stirred for 35 hours at 75-800C. The reaction mixture was cooled to 25-350C and stirred for 45 minutes at 25-35°C. The solvent was distilled off under reduced pressure. Water (100 ml) was added to the obtained residue. The reaction mixture was cooled to 25-350C. The reaction mixture was extracted with ethyl acetate. The solvent was distilled off to get the title compound as a residue Yield: lgram. Example-6:
Preparation of condensed compound of formula-4d:
A mixture of 20 grams of l-cyclopropyl-6,7-difluoro-8-methoxy-l,4-dihydro-4- oxoquinoline-3-carboxamide, 2.9 grams of (S,S)-2,8-diazabicyclo (4.3.0) nonane , 100 ml of acetonitrile and 2 grams of diazabicyclo[5.4.0]undec-7-ene (DBU) was heated to 75-80°C. The reaction mixture was stirred for 35 hours at 75-80°C. The reaction mixture was cooled to 25-350C. The reaction mixture was stirred for 45 minutes at 25-350C. The solid obtained was filtered and washed with acetonitrile. The material was dried at 40-450C to get the title compound. Yield: 12 grams; M.R: 210-2120C.
Example-7:
Preparation of condensed compound of formula-4e:
A mixture of (S,S)-8-trityl-2,8-diazabicyclo (4.3.0) nonane (42 grams), l-cyclopropyl-N,N-diethyl-6,7-difluoro-l,4-dihydro-8-methoxy-4-oxoquinoline-3- carboxamide (10 grams), potassium carbonate ( 7.9 grams) and dimethyl formamide (80 ml) was heated to 110-1200C and stirred for 20 hours at 110-1200C. The reaction mixture was cooled to 800C. The solvent was distilled off under reduced pressure. Water (100 ml) was added to the obtained residue. The reaction mixture was cooled to 25-35°C. The reaction mixture was extracted with ethyl acetate. The solvent was distilled off to obtain the title compound as a residue. Yield: 14.5 grams
Example-8: Preparation of moxifloxacin from 4d:
A mixture of 165 ml of water, 35 grams of sodium hydroxide, 150 ml of ethylene glycol and 12 grams of condensed compound of formula-4d was heated to 115°C. The reaction mixture was stirred for 15 hours at 115°C. The reaction mixture was cooled to 5°C. The pH of the reaction mixture was adjusted to 5.5 using hydrochloric acid and stirred for 30 minutes at 5°C. The obtained solid was filtered and washed with water. The solid was dried at 45°C to get the title compound. Yield: 8 grams; M.R: 203-2050C Example-9:
Preparation of moxifloxacin from 4b:
Moxifloxacin can be prepared from 4b (3grams), by a method which is analogous to the method illustrated in Example-8 Yield: 0.85 grams; M.R: 203-205°C
Example-10:
Preparation of moxifloxacin from 4c:
Moxifloxacin can be prepared from 4c (3 grams), by a method which is analogous to the method illustrated in Example-8 Yield: 1.0 grams; M.R: 203-205°C
Example-11:
Preparation of moxifloxacin from 4a: Moxifloxacin can be prepared from 4a (10 grams), by a method which is analogous to the method illustrated in Example-8 Yield: 7.5 grams; M.R: 203-2050C
Example-12: Preparation of moxifloxacin from 4e:
The condensed compound of formula-4e (14.5 grams) was dissolved in ethyl acetate 100ml. Aqueous hydrochloric acid (5 ml in 20 ml of water) was added to the above reaction mixture. The reaction mixture was stirred at 25-30°C for 30-45 min. 20ml of water was added to the reaction mixture. The two layers were separated. The aqueous layer was washed twice with ethyl acetate. The pH of the aqueous layer was adjusted to 10.8 using sodium hydroxide solution. The reaction mixture was extracted with methylene chloride. The solvent distilled off to get a residue. The residue was suspended in 165 ml of water, 35 grams of sodium hydroxide; 150 ml of ethylene glycol was heated to 115°C. The reaction mixture was stirred for 15 hours at 115°C. The reaction mixture was cooled to 5°C. The pH of the reaction mixture was adjusted to 5.5 using hydrochloric acid and stirred for 30 minutes at 5°C.The obtained solid was filtered and washed with water and dried at 450C to get the title compound. Yield: 4.5 grams; M.R: 203-2050C
Example-13:
Preparation of moxifloxacin hydrochloride compound of formula-1:
A mixture of 8 grams of moxifloxacin, 16 ml of water and 64 ml of methanol was heated to reflux temperature of 700C. The reaction mixture was filtered to remove undissolved material. Filtrate was cooled to 35°C. The pH of the filtrate was adjusted to 1.6 using hydrochloric acid. The reaction mixture was cooled to 150C. The reaction mixture was stirred for 45 minutes at 150C. The solid obtained was filtered and washed with methanol and dried at 40-450C to get crystalline Form-Y of moxifloxacin hydrochloride monohydrate. Yield: 5.5 grams
Example- 14:
Preparation of moxifloxacin hydrochloride monohydrate:
A mixture of 25 grams of moxifloxacin, 16 ml of water and 64 ml of methanol was heated to reflux temperature of 700C. The reaction mixture was filtered to remove undissolved material. Filtrate was cooled to 35°C. The pH of the filtrate was adjusted to 1.6 using hydrochloric acid. The reaction mixture was cooled to 150C. The reaction mixture was stirred for 45 minutes at 150C. The obtained solid was taken into a mixture of 22 ml of water and 0.5ml of hydrochloric acid and stirred 30 min at 5°C. The compound was filtered and washed with water, dried at 40-450C to get moxifloxacin hydrochloride monohydrate particles having oval shape. Yield: 20 grams
Example-15:
Preparation of anhydrous crystalline Form-I of moxifloxacin hydrochloride: Moxifloxacin hydrochloride (Form-Y, 10 grams) suspended in 50 ml of methanol and the reaction mixture was heated to 45-5O0C. 18ml of dichloromethane was added slowly to the reaction mixture. The reaction mixture was stirred at 45-50°C for 15-20 minutes. The reaction mixture was cooled slowly to around 0-5°C and stirred for 1-1.5 hours. The precipitated solid was filtered under nitrogen atmosphere and washed with 5 ml of methanol. The solid obtained was dried at 110-120°C till the moisture content reached below 0.5%. Yield: 8.0 grams.
Bulk density: 0.28 g/ml; Tapped density: 0.52 g/ml Particle size distribution : D( v,0.1) :1.6 μm ; D( vs0.5) : 5.5 μm ; D( v,0.9) :25.0 μm
……………………….
SYNTHESIS
Preparation of Moxifloxacin
A solution of lOOg of 1-Cyclopropyl -6,7difluoro-l,4-dihydro-4-oxo-methoxyquinolone- 3-carboxilic acid), 52gr of (S,S)2,8 diazabicyclo[4,3,0]nonane, lOgr of 1,8- diazabycyclo[5,4,0]undec-7-ene and 300ml of acetonitrile is heated to 65 -70°C temperature and stirred till the reaction get completed and evaporated the solvent. Added 5 %aqueous isopropylalcohol to the reaction mass and adjusted the pH to basic with caustic solution and then filtered the reaction mass. Combined the total filtrate and adjusted the pH to 5.0 to 6.0 with aqueous HCl.and isolated the separated solid at a temperature of 10-150C to afford Moxifloxacin.
………………….
SYNTHESIS REVIEW
Beilstein J. Org. Chem. 2013, 9, 2265–2319.
While isolated pyridines are a vital component in numerous drugs the related quinoline/quinolone scaffolds are becoming increasingly common. One such example is nalidixic acid (
1.101, in fact a naphthyridone,
Figure 2). Nalidixic acid is a prototype quinolone antibiotic that has been used extensively as an effective treatment against both gram positive and gram negative bacteria. In general, quinolone antibiotics act by interfering with the enzymes DNA-gyrase and/or topoisomerase of bacteria
[55]. Moxifloxacin (
1.102, Avelox) and levofloxacin (
1.103, Levaquin) are two third-generation fluoroquinolone antibiotics which also appear amongst the top selling drugs. These compounds display similar SAR data
[56]. For instance, in the case of moxifloxacin the fluorine atom in the C6 position enhances microbial activity while a methoxy group in the C8 position is reported to increase potency and decrease toxicity. Furthermore, it was found that a cyclopropyl group was beneficial for the enzyme–DNA binding complex while the bulky nitrogen-based appendage at C7 helps to bind to DNA gyrase and hinders the efflux of the drug from the bacterial cell.
![[1860-5397-9-265-2]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-2.png?max-width=550&background=EEEEEE)
Figure 2: Structures of nalidixic acid, levofloxacin and moxifloxacin.
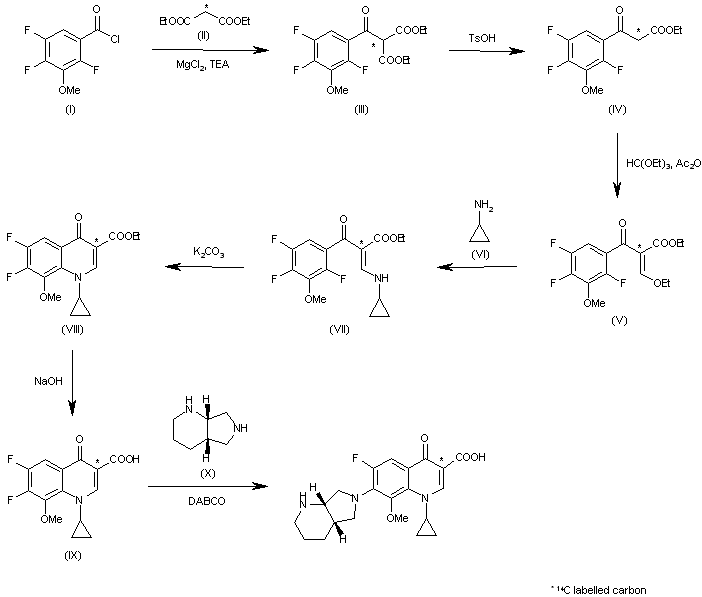
Due to the elaborate substitution pattern of the parent quinolone ring systems these compounds are usually prepared via a linear consecutive sequence. In the case of moxifloxacin, an intramolecular base catalysed nucleophilic aromatic substitution is used to prepare the bicyclic ring system of the highly substituted aromatic
1.104 (
Scheme 19). A SNAr reaction is then used to introduce the saturated piperidinopyrrolidine appendage
1.105to furnish the desired structure
[57-60]. In order to obtain a high yield for the substitution reaction a one-pot procedure was developed, initial masking of the acid (
1.104) is achieved by silylation with subsequent borane chelate formation. Addition of the amine nucleophile
1.105 under basic conditions then renders the desired product in high yield. The available patent literature however does not comment on regioselectivity issues of the SNAr reaction due to the presence of the second fluoride substituent in the substrate, although not necessarily as electronically favourable for displacement it is certainly more accessible.
![[1860-5397-9-265-i19]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i19.png?max-width=550&background=EEEEEE)
The saturated (
S,
S)-2,8-diazabicyclo[4.3.0]nonane (
1.105) used in the final step can be prepared by a double nucleophilic substitution between tosylamine and 2,3-
bis-chloromethylpyridine (
1.112) followed by catalytic reduction of the resulting bicycle using palladium on carbon in acetic acid (
Scheme 20). As the corresponding sulfonamide
1.113 was found to be a crystalline solid a resolution using (D)-(+)-
O,
O-dibenzoyltartaric acid was reported to separate the enantiomers
[Petersen, U.; Schenke, T.; Krebs, A.; Schenke, T.; Philipps, T.; Grohe, K.; Bremm, K.-D.; Endermann, R.; Metzger, K. G. New Quinoline and Naphthyridinonecarboxylic Acid Derivatives. Ger. Patent DE 4 208 792 A1, March 23, 1993.].
Scheme 20: Synthesis of (
S,
S)-2,8-diazabicyclo[4.3.0]nonane
1.105.
…………………………………
PAPER
First way of enantioselective synthesis of moxifloxacin intermediate
LI GuangXun, WU Lei, FU QingQuan, TANG Zhuo, ZHANG XiaoMei
A new method of enantioselective synthesis of (S,S)-2,8-diazobicyclo [4.3.0] nonane was found by using (R)-2-amino-2-phenyl-ethanol as chiral induction reagent. The entire synthetic process included 8 steps which were easy to operate with high yield. The purification method was only simple recrystallization or even used directly in the next step without further purification. The total yield was 29%.
|
| 2013 Vol. 56 (3): 307-311 [Abstract] ( 22 ) [ PDF (518 KB) ] ( 92 ) [Supporting Information] DOI: 10.1007/s11426-012-4803-7 |
- Drugwatch. “Avelox Patent Family”. Retrieved 16 September 2012.
- “Details for NDA:021085″. DrugPatentWatch. Retrieved 17 July 2009.
- “www.accessdata.fda.gov”.
- “www.emea.europa.eu”.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021085s047,021277s041lbl.pdf
- “Avelox”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/1999/21085ltr.pdf
- http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21-085S010_Avelox_Approv.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2004/21085se1-022,21277se1-017ltr.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/021085s026,021277s022ltr.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/021085s027,029,021277s024,025ltr.pdf.
- “Moxifloxacin: restricted use : MHRA”.
- European Medicines Agency (24 July 2008). “European Medicines Agency recommends restricting the use of oral moxifloxacin-containing medicines”. Retrieved 20 July 2009.
- “SYNOPSIS”. Retrieved 29 January 2009.
- Karande SC, Kshirsagar NA (February 1992). “Adverse drug reaction monitoring of ciprofloxacin in pediatric practice”. Indian Pediatr 29 (2): 181–8. PMID 1592498.
- Dolui SK, Das M, Hazra A (2007). “Ofloxacin-induced reversible arthropathy in a child”. Journal of Postgraduate Medicine 53 (2): 144–5. doi:10.4103/0022-3859.32220 .PMID 17495385.
- “Center for drug evaluation and research Application number 21-598″. Food and Drug Administration (FDA). 15 April 2005. Retrieved 21 July 2009.
- Jump up^ Babar, S. (October 2013). “SIADH Associated With Ciprofloxacin.”. The Annals of Pharmacotherapy (Sage Publiahing) 47 (10): 1359–1363.doi:10.1177/1060028013502457 . ISSN 1060-0280.PMID 24259701. Retrieved November 139,2013.
- Renata Albrecht (28 July 2004). “NDA 21-085/S-024, NDA 21-277/S-019″. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Renata Albrecht (31 May 2007). “NDA 21-085/S-036, NDA 21-277/S-030″. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Renata Albrecht (15 February 2008). “NDA 21-085/S-038, NDA 21-277/S-031″. Division of Special Pathogen and Transplant Products. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- DEPARTMENT OF HEALTH & HUMAN SERVICES (28 February 2008). “NDA 21-085/S-014, S-015, S-017″. Food and Drug Administration (FDA). Retrieved 17 July 2009.
- Nea Zealand Government (26 June 2003). “Adverse Reaction Reporting and IMMP”. New Zealand: Medsafe. Retrieved 30 January 2009.
- http://www.mhra.gov.uk/Safetyinformation/DrugSafetyUpdate/CON087781, retrieved 2012-11-10 Missing or empty
|title= (help)
- http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf, retrieved 2012-11-10 Missing or empty
|title= (help)
- “EU agency recommends restricting moxifloxacin use”. Reuters. 24 July 2008. Retrieved 21 July 2009.
- Renata Albrecht (3 October 2008). “NDA 21-085/S-040, NDA 21-277/S-034″. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Updated Labeling for Antibiotic Avelox (Moxifloxacin) Regarding Risk of Severe Liver Injury / Information Update: 2010-42 For immediate release 22 March 2010http://www.hc-sc.gc.ca/ahc-asc/media/advisories-avis/_2010/2010_42-eng.php
- Saint F, Gueguen G, Biserte J, Fontaine C, Mazeman E (September 2000). “[Rupture of the patellar ligament one month after treatment with fluoroquinolone]“. Rev Chir Orthop Reparatrice Appar Mot (in French) 86 (5): 495–7.PMID 10970974.
- http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf, retrieved 2012-11-10 Missing or empty
|title= (help)
- Bayer (December 2008). “AVELOX (moxifloxacin hydrochloride) Tablets AVELOX I.V. (moxifloxacin hydrochloride in sodium chloride injection)”. Food and Drug Administration (FDA). p. 19. Retrieved 2 November 2010. Unknown parameter
|line= ignored (help)
- “Moxifloxacin”. University of Maryland Medical Center. 2009. Retrieved 22 July 2009.
- http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf
- Shin HC, Kim JC, Chung MK (September 2003). “Fetal and maternal tissue distribution of the new fluoroquinolone DW-116 in pregnant rats”. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 136 (1): 95–102.doi:10.1016/j.cca.2003.08.004 . PMID 14522602.
- Dan M, Weidekamm E, Sagiv R, Portmann R, Zakut H (February 1993). “Penetration of fleroxacin into breast milk and pharmacokinetics in lactating women” (PDF).Antimicrob. Agents Chemother. 37 (2): 293–6.doi:10.1128/AAC.37.2.293 . PMC 187655.PMID 8452360.
- Noel GJ, Bradley JS, Kauffman RE (October 2007). “Comparative safety profile of levofloxacin in 2523 children with a focus on four specific musculoskeletal disorders”.Pediatr. Infect. Dis. J. 26 (10): 879–91.doi:10.1097/INF.0b013e3180cbd382 . PMID 17901792.
- Joette Meyer; Renata Albrecht (16 March 2004).“Division of Special Pathogen and Immunologic Drug Products Summary of Clinical Review of Studies Submitted in Response to a Pediatric Written Request”. Food and Drug Administration (FDA). Retrieved 22 July 2009.
- Elbe DH, Chang SW (February 2005). “Moxifloxacin-warfarin interaction: a series of five case reports”. Ann Pharmacother 39 (2): 361–4. doi:10.1345/aph.1E179 .PMID 15632222.
- Karen E. Brown. “Top Ten Dangerous Drug Interactions in Long-Term Care”. Medication Management Project. Retrieved 1 August 2009.
- Stass HH (29 June – 3 July 1997). “Bay 12-8039 (BA) does not interact with theophylline (TH)”. Presented at the 20th International Congress of Chemotherapy.
- Medscape: Medscape Access
- http://69.20.19.211/cder/warn/dec99/wl121599.pdf
- Renata Albrecht (16 May 2002). “NDA 21-085/S-012″.Food and Drug Administration (FDA). Retrieved 17 July 2009.
- Drlica K, Zhao X (1 September 1997). “DNA gyrase, topoisomerase IV, and the 4-quinolones”. Microbiol Mol Biol Rev. 61 (3): 377–92. PMC 232616. PMID 9293187.
- “Drug card for Moxifloxacin (DB00218)”. Canada: DrugBank. 19 February 2009. Retrieved 3 August 2009.
- Alffenaar J. W. C., van Altena R., Bökkerink H. J (2009). “Pharmacokinetics of moxifloxacin in cerebrospinal fluid and plasma in patients with tuberculous meningitis”. Clinical Infectious Diseases 49 (7): 1080–2. doi:10.1086/605576 .PMID 19712035.
- “Inventors/Applicants”. patentlens.net. 3 October 2006. Retrieved 17 July 2009.
- Ed Lamb (1 May 2008). “Top 200 Prescription Drugs of 2007″. Pharmacy Times. Retrieved 21 July 2009.
- http://www.uspto.gov/web/offices/pac/dapp/opla/term/certs/4990517.pdf
- US 4990517 A (Feb 1991) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+4990517#list US 5051509 A (Sep 1991) Nagano et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5051509#list US 5059597 A (Oct 1991) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5059597#list US 5395944 A (Mar 1995) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5395944#list US 5416096 A (May 1995) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5416096#list
- Renata Albrecht (12 June 2002). “NDA 21-085/S-006, S-007 – NDA 21-277/S-002″. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- The 114Th Medicines Adverse Reactions Committee Meeting; Professor P. Ellis, Professor D.C.G. Skegg, Dr M. Rademaker, Dr F. McClure, Dr N. Rafter, Dr M. Tatley (26 June 2003). “Adverse Reaction Reporting and IMMP”. New Zealand: Medsafe. Retrieved 12 August 2009.
- Renata Albrecht, (6 October 2003). “NDA 21-085/S-019 – NDA 21-277/S-011″. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- Renata Albrecht (6 October 2003). “NDA 21-085/S-039 NDA 21-277/S-033″. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- Bayer HealthCare Pharmaceuticals Inc. (August 2008).“MEDICATION GUIDE AVELOX (AV-eh-locks) (moxifloxacin hydrochloride) Tablets AVELOX I.V. (AV-eh-locks) (moxifloxacin hydrochloride in sodium chloride injection)”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- Ozlem Belen (24 June 2009). “NDA 21-085/S-041 NDA 21-277/S-035″. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- Bailey RR, Natale R, Linton AL (October 1972). “Nalidixic acid arthralgia”. Can Med Assoc J 107 (7): 604 passim.PMC 1940945. PMID 4541768.
- Bailey RR, Kirk JA, Peddie BA (July 1983). “Norfloxacin-induced rheumatic disease”. N Z Med J 96 (736): 590.PMID 6223241.
- Szarfman A, Chen M, Blum MD (January 1995). “More on fluoroquinolone antibiotics and tendon rupture” (PDF). N Engl J Med 332 (3): 193.doi:10.1056/NEJM199501193320319 . PMID 7800023.
- “Petition to Require a Warning on All Fluoroquinolone Antibiotics (HRG Publication #1399)”. Public Citizen. 1 August 1996. Retrieved on 27 December 2008.
- “Reports of adverse events with fluoroquinolones”. FDA Medical Bulletin 26 (3). October 1996.[dead link] Retrieved on 27 December 2008.
- “Madigan, Public Citizen, petition FDA for “black box” warning regarding potential adverse effects of certain popular antibiotics” (Press release). Office of the Illinois Attorney General. 29 August 2006. Retrieved 27 December 2008.
- “Public Citizen Petitions the FDA to Include a Black Box Warning on Fluoroquinolone Antibiotics (HRG Publication #1781)”. Public Citizen. 29 August 2006. Retrieved 27 December 2008.
- “Public Citizen v. Food and Drug Administration (FDA) (Fluoroquinolone)”. Public Citizen. 3 January 2008. Retrieved 27 December 2008.
- Ravn, Karen (18 August 2008). “Behind the FDA’s ‘black box’ warnings”. Los Angeles Times. Retrieved 27 December 2008.
- “FDA Requests Boxed Warnings on Fluoroquinolone Antimicrobial Drugs” (Press release). U.S. Food and Drug Administration. 8 July 2008. Retrieved 11 October 2008.
- “Drugs@FDA”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- “MedWatch: The FDA Safety Information and Adverse Event Reporting Program”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- MacCarthy, Paul (22 October 2008). “Important Change in the Avelox (moxifloxacin hydrochloride) and Cipro (ciprofloxacin) Complete Prescribing Information – Addition of Boxed Warning and Medication Guide Regarding Tendinitis and Tendon Rupture”. Bayer HealthCare Pharmaceuticals. Retrieved 27 December 2008.
- Bayer AG (6 November 2007). “Ruling in Bayer’s favor over Avelox patents”. Bayer. Retrieved 29 August 2009.
- http://www.orangebookblog.com/Bayer_20v._20Dr._20Reddy_27s.pdf
- “United States Court of Appeals for the Federal Circuit”. uscourts.gov. 28 June 2007. Retrieved 29 August 2009.
- Bayer AG (24 April 2008). “Risk Report”. Bayer. Retrieved 29 August 2009.
- In The United States District Court For The District Of Columbia Public Citizen, Inc. VS. Food And Drug Administration 3 January 2008
- Office of the Attorney General State of Illinois Lisa Madigan Citizen Petition to Include a Black Box Warning on Fluoroquinolone Antibiotics 18 May 2005
- Public Citizen’s Petition to Include a Black Box Warning on Fluoroquinolone Antibiotics (HRG Publication #1781) 29 August 2006
- Public Citizen’s Petition to Require a Warning on All Fluoroquinolone Antibiotics (HRG Publication #1399) 1 August 1996
- Public Citizen’s Petition to Ban the Antibiotic Gatifloxacin (Tequin) (HRG Publication #1768)
- Public Citizen’s Petition to immediately ban the antibiotic Trovafloxacin (Trovan). (HRG Publication #1485) Date: 3 June 1999
- Public Citizen’s Petition to immediately stop the distribution of dangerous, misleading prescription drug information to the public. HRG Publication #1442 Date: 9 June 1998
- June 2004, A petition To the United States Congress to immediately take action to protect consumers from the reckless and negligent abuses of the FDA and the following Pharmaceutical Companies: Bayer, Ortho-McNeill, Pfizer, Merck, Bristol-Myers Squibb, Sanofi Winthrop, Bertek Pharmaceuticals – Rhone-Poulenc Rorer and Barr. These companies manufacture and distribute fluoroquinolone antibiotics in the United States in a manner that fails to warn of serious adverse event risks, and downplays and fails to warn physicians of the serious risks associated with fluoroquinolone therapy.
| EP0350733A2 | Jun 30, 1989 | Jan 17, 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 | Dec 28, 1992 | Jul 14, 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0591808A1 | Sep 27, 1993 | Apr 13, 1994 | Bayer Ag | Quinolonecarboxylic acids |
| EP0592868A1 | Sep 29, 1993 | Apr 20, 1994 | Bayer Ag | Quinolonecarboxylic acids |
| US5849752 | Dec 5, 1996 | Dec 15, 1998 | Bayer Aktiengesellschaft | Crystal modification of CDCH a process for its preparation and pharmaceutical formulations comprising this modification |
| WO1999026940A2 | Nov 12, 1998 | Jun 3, 1999 | Bayer Ag | Method for producing 8-methoxy-quinoline carboxylic acids |
| WO2004091619A1 | Apr 9, 2004 | Oct 28, 2004 | Reddys Lab Ltd Dr | A crystalline form iii of anhydrous moxifloxacin hydrochloride and a process for preparation thereof |
| WO2007010555A2 | Jul 13, 2006 | Jan 25, 2007 | Ramprasad Achampeta Kodanda | Novel crystalline forms of moxifloxacin hydrochloride and process for preparation thereof |
| WO2008059521A2 | Sep 27, 2007 | May 22, 2008 | Ramprasad Achampeta Kodanda | Novel process for the preparation of moxifloxacin hydrochloride and a novel polymorph of moxifloxacin |
| CN102030751B | Dec 1, 2010 | Nov 21, 2012 | 上虞京新药业有限公司 | Process for crystallizing moxifloxacin hydrochloride |
| EP2154137A1 | Aug 4, 2008 | Feb 17, 2010 | Chemo Ibérica, S.A. | Crystalline form of moxifloxacin base |
| US20110212990 * | Nov 6, 2008 | Sep 1, 2011 | Hetero Research Foundation | Novel polymorph of moxifloxacin hydrochloride |
| WO2005012285A1 * | Aug 5, 2004 | Feb 10, 2005 | Satyanarayana Chava | An improved process for the preparation of moxifloxacin hydrochloride |
| EP0443498A1 * | Feb 18, 1991 | Aug 28, 1991 | Kyorin Pharmaceutical Co., Ltd. | Isoindoline derivatives |
| EP0550903A1 * | Dec 28, 1992 | Jul 14, 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| US6323213 * | Feb 12, 1997 | Nov 27, 2001 | Bayer Aktiengesellschaft | Possibly substituted 8-cyano-1-cyclopropyl-7-(2,8-diazabicyclo-[4.3.0]-nonan-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinolin carboxylic acids and their derivatives |
| US5849752 * | Dec 5, 1996 | Dec 15, 1998 | Bayer Aktiengesellschaft | Crystal modification of CDCH a process for its preparation and pharmaceutical formulations comprising this modification |
| WO2007010555A2 * | Jul 13, 2006 | Jan 25, 2007 | Ramprasad Achampeta Kodanda | Novel crystalline forms of moxifloxacin hydrochloride and process for preparation thereof |
| WO2008059521A2 * | Sep 27, 2007 | May 22, 2008 | Ramprasad Achampeta Kodanda | Novel process for the preparation of moxifloxacin hydrochloride and a novel polymorph of moxifloxacin |
| WO2008138759A1 * | Apr 30, 2008 | Nov 20, 2008 | Sandoz Ag | Process for the preparation of moxifloxacin hydrochloride |
| WO2007148137A1 * | Jun 22, 2007 | Dec 27, 2007 | Generics Uk Ltd | Novel hydrate form of moxifloxacin monohydrochloride |
| WO2008028959A1 * | Sep 7, 2007 | Mar 13, 2008 | Sint Quimica Sa | Crystalline form of moxifloxacin hydrochloride |
| WO2008059223A2 * | Nov 13, 2007 | May 22, 2008 | Cipla Ltd | Process for the synthesis of moxifloxacin hydrochloride |
| WO2008095964A1 * | Feb 6, 2008 | Aug 14, 2008 | Sint Quimica Sa | Crystalline form of moxifloxacin base |
| WO2009087151A1 * | Jan 7, 2009 | Jul 16, 2009 | Chemo Iberica Sa | Polymorphic forms of moxifloxacin hydrochloride and processes for preparation thereof |
| CN102603738B | Feb 24, 2012 | Dec 11, 2013 | 天津市汉康医药生物技术有限公司 | 一种稳定的盐酸莫西沙星化合物 |
| EP2083010A1 | Jan 8, 2008 | Jul 29, 2009 | Chemo Ibérica, S.A. | Polymorphic Forms of Moxifloxacin hydrochloride and processes for preparation thereof |
| EP2154137A1 | Aug 4, 2008 | Feb 17, 2010 | Chemo Ibérica, S.A. | Crystalline form of moxifloxacin base |
| US8198451 | Nov 13, 2007 | Jun 12, 2012 | Cipla Limited | Process for the synthesis of moxifloxacin hydrochloride |
| US20110212990 * | Nov 6, 2008 | Sep 1, 2011 | Hetero Research Foundation | Novel polymorph of moxifloxacin hydrochloride |

 KW 4490
KW 4490



























![[1860-5397-9-265-2]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-2.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-265-i19]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i19.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-265-i20]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i20.png?scale=2.0&max-width=1024&background=FFFFFF)
