CS-3150, (XL550)
CS 3150, angiotensin II receptor antagonist, for the treatment or prevention of such hypertension and heart disease similar to olmesartan , losartan, candesartan , valsartan, irbesartan, telmisartan, eprosartan,
Cas name 1H-Pyrrole-3-carboxamide, 1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-, (5S)-
CAS 1632006-28-0 for S conf
MF C22 H21 F3 N2 O4 S
MW 466.47
(S)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
CAS 1632006-28-0 for S configuration
1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide
(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide, CAS 880780-76-7
(+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-82-4
(-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide..1072195-83-5
WO2008 / 126831 (US Publication US2010-0093826)http://www.google.co.in/patents/EP2133330A1?cl=en
WO 2006012642..compound A;..http://www.google.com/patents/WO2006012642A2?cl=en
WO2006 / 012642 (US Publication US2008-0234270)
- Originator Exelixis
- Developer Daiichi Sankyo Company..
Daiichi Sankyo Company,Limited, 第一三共株式会社 - Class Antihypertensives; Small molecules
- Mechanism of Action Mineralocorticoid receptor antagonists

JAPAN PHASE 2……….Phase 2 Study to Evaluate Efficacy and Safety of CS-3150 in Patients with Essential Hypertension
Phase II Diabetic nephropathies; Hypertension
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Diabetic nephropathies in Japan (NCT02345057)
- 01 Jan 2015 Daiichi Sankyo initiates a phase IIb trial for Hypertension in Japan (NCT02345044)
- 01 May 2013 Phase-II clinical trials in Diabetic nephropathies in Japan (PO)
- Currently, angiotensin II receptor antagonists and calcium antagonists are widely used as a medicament for the treatment or prevention of such hypertension or heart disease.Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.Renin – angiotensin II receptor antagonists are inhibitors of angiotensin system is particularly effective in renin-dependent hypertension, and show a protective effect against cardiovascular and renal failure. Also, the calcium antagonists, and by the function of the calcium channel antagonizes (inhibits), since it has a natriuretic action in addition to the vasodilating action, is effective for hypertension fluid retention properties (renin-independent) .Therefore, the MR antagonist, when combined angiotensin II receptor antagonists or calcium antagonists, it is possible to suppress the genesis of multiple hypertension simultaneously, therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology is expected to exhibit.Also, diuretics are widely used as a medicament for the treatment or prevention of such hypertension or heart disease. Diuretic agent is effective in the treatment of hypertension from its diuretic effect. Therefore, if used in combination MR antagonists and diuretics, the diuretic effect of diuretics, it is possible to suppress the genesis of multiple blood pressure at the same time, shows a therapeutic or prophylactic effect of the stable and sufficient hypertension irrespective of the etiology it is expected.1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (hereinafter, compound ( I)) is, it is disclosed in Patent Documents 1 and 2, hypertension, for the treatment of such diabetic nephropathy are known to be useful.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Useful as a mineralocorticoid receptor (MR) antagonist, for treating hypertension, cardiac failure and diabetic nephropathy. It is likely to be CS-3150, a non-steroidal MR antagonist, being developed by Daiichi Sankyo (formerly Sankyo), under license from Exelixis, for treating hypertension and diabetic nephropathy (phase 2 clinical, as of March 2015). In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month (NCT02345057).
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.


Mineralocorticoid receptor (MR) (aldosterone receptor) has been known to play an important role in the control of body electrolyte balance and blood pressure, spironolactone having a steroid structure, MR antagonists such as eplerenone, are known to be useful in the treatment of hypertension-heart failure.
CS-3150 (XL550) is a small-molecule antagonist of the mineralocorticoid receptor (MR), a nuclear hormone receptor implicated in a variety of cardiovascular and metabolic diseases. MR antagonists can be used to treat hypertension and congestive heart failure due to their vascular protective effects. Recent studies have also shown beneficial effects of adding MR antagonists to the treatment regimen for Type II diabetic patients with nephropathy. CS-3150 is a non-steroidal, selective MR antagonist that has the potential for the treatment of hypertension, congestive heart failure, or end organ protection due to vascular damage.
Exelixis discovered CS-3150 and out-licensed the compound to Daiichi-Sankyo. Two phase 2a clinical trials, one in hypertensive patients and the other in type 2 diabetes with albuminuria, are currently being conducted in Japan by Daiichi-Sankyo.
Daiichi Sankyo (formerly Sankyo), under license from Exelixis, is developing CS-3150 (XL-550), a non-steroidal mineralocorticoid receptor (MR) antagonist, for the potential oral treatment of hypertension and diabetic nephropathy, microalbuminuria , By October 2012, phase II development had begun ; in May 2014, the drug was listed as being in phase IIb development . In January 2015, a phase II trial for type 2 diabetes mellitus and microalbuminuria was planned to be initiated later that month. At that time, the trial was expected to complete in March 2017 .
Exelixis, following its acquisition of X-Ceptor Therapeutics in October 2004 , was investigating the agent for the potential treatment of metabolic disorders and cardiovascular diseases, such as hypertension and congestive heart failure . In September 2004, Exelixis expected to file an IND in 2006. However, it appears that the company had fully outlicensed the agent to Sankyo since March 2006 .
| Description | Small molecule antagonist of the mineralocorticoid receptor (MR) |
| Molecular Target | Mineralocorticoid receptor |
| Mechanism of Action | Mineralocorticoid receptor antagonist |
| Therapeutic Modality | Small molecule |
In January 2015, a multi-center, placebo-controlled, randomized, 5-parallel group, double-blind, phase II trial (JapicCTI-152774; NCT02345057; CS3150-B-J204) was planned to be initiated later that month in Japan, in patients with type 2 diabetes mellitus and microalbuminuria, to assess the efficacy and safety of different doses of CS-3150 compared to placebo. At that time, the trial was expected to complete in March 2017; later that month, the trial was initiated in the Japan
By October 2012, phase II development had begun in patients with essential hypertension
By January 2011, phase I trials had commenced in Japan
Several patents WO-2014168103,
WO-2015012205 and WO-2015030010
XL-550, claimed in WO-2006012642,
………………………………………………………………….
(Example 3)(+/-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
- After methyl 4-methyl-5-[2-(trifluoromethyl) phenyl]-1H-pyrrole-3-carboxylate was obtained by the method described in Example 16 of WO 2006/012642 , the following reaction was performed using this compound as a raw material.
- Methyl 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate (1.4 g, 4.9 mmol) was dissolved in methanol (12 mL), and a 5 M aqueous sodium hydroxide solution (10 mL) was added thereto, and the resulting mixture was heated under reflux for 3 hours. After the mixture was cooled to room temperature, formic acid (5 mL) was added thereto to stop the reaction. After the mixture was concentrated under reduced pressure, water (10 mL) was added thereto to suspend the resulting residue. The precipitated solid was collected by filtration and washed 3 times with water. The obtained solid was dried under reduced pressure, whereby 4-methyl-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylic acid (1.1 g, 83%) was obtained as a solid. The thus obtained solid was suspended in dichloromethane (10 mL), oxalyl chloride (0.86 mL, 10 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 2 hours. After the mixture was concentrated under reduced pressure, the residue was dissolved in tetrahydrofuran (10 mL), and 4-(methylsulfonyl)aniline hydrochloride (1.0 g, 4.9 mmol) and N,N-diisopropylethylamine (2.8 mL, 16 mmol) were sequentially added to the solution, and the resulting mixture was heated under reflux for 18 hours. After the mixture was cooled to room temperature, the solvent was distilled off under reduced pressure, and acetonitrile (10 mL) and 3 M hydrochloric acid (100 mL) were added to the residue. A precipitated solid was triturated, collected by filtration and washed with water, and then, dried under reduced pressure, whereby 4-methyl-N-[4-(methylsulfonyl) phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (1.4 g, 89%) was obtained as a solid.
1H-NMR (400 MHz, DMSO-d6) δ11.34 (1H, brs,), 9.89 (1H, s), 7.97 (2H, d, J = 6.6 Hz), 7.87-7.81 (3H, m), 7.73 (1H, t, J = 7.4 Hz), 7.65-7.61 (2H, m), 7.44 (1H, d, J = 7.8 Hz), 3.15 (3H, s), 2.01 (3H, s). - Sodium hydride (0.12 g, 3 mmol, 60% dispersion in mineral oil) was dissolved in N,N-dimethylformamide (1.5 mL), and 4-methyl -N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide (0.47 g, 1.1 mmol) was added thereto, and then, the resulting mixture was stirred at room temperature for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (0.14 g, 1.2 mmol) was added thereto, and the resulting mixture was stirred at room temperature. After 1 hour, sodium hydride (40 mg, 1.0 mmol, oily, 60%) was added thereto again, and the resulting mixture was stirred for 30 minutes. Then, 1,3,2-dioxathiolane-2,2-dioxide (12 mg, 0.11 mmol) was added thereto, and the resulting mixture was stirred at room temperature for 1 hour. After the mixture was concentrated under reduced pressure, methanol (5 mL) was added to the residue and insoluble substances were removed by filtration, and the filtrate was concentrated again. To the residue, tetrahydrofuran (2 mL) and 6 M hydrochloric acid (2 mL) were added, and the resulting mixture was stirred at 60°C for 16 hours. The reaction was cooled to room temperature, and then dissolved in ethyl acetate, and washed with water and saturated saline. The organic layer was dried over anhydrous sodium sulfate and filtered. Then, the filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate), whereby the objective compound (0.25 g, 48%) was obtained.
1H-NMR (400 MHz, CDCl3) δ: 7.89-7.79 (m, 6H), 7.66-7.58 (m, 2H), 7.49 (s, 1H), 7.36 (d, 1H, J = 7.4Hz), 3.81-3.63 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1246.
Anal. calcd for C22H21F3N2O4S: C, 56.65; H, 4.54; N, 6.01; F, 12.22; S, 6.87. found: C, 56.39; H, 4.58; N, 5.99; F, 12.72; S, 6.92.
(Example 4)
Optical Resolution of Compound of Example 3

 OLMESARTAN
OLMESARTAN

- Resolution was performed 4 times in the same manner as in Example 2, whereby 74 mg of Isomer C was obtained as a solid from a fraction containing Isomer C (tR = 10 min), and 71 mg of Isomer D was obtained as a solid from a fraction containing Isomer D (tR = 11 min).
- Isomer C: (+)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: +7.1° (c = 1.0, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.91 (s, 1H), 7.87-7.79 (m, 5H), 7.67-7.58 (m, 2H), 7.51 (s, 1H), 7.35 (d, 1H, J = 7.0 Hz), 3.78-3.65 (m, 4H), 3.05 (s, 3H), 2.07 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1260.
Retention time: 4.0 min. - Isomer D: (-)-1-(2-hydroxyethyl)-4-methyl-N-[4-(methylsulfonyl)phenyl]-5-[2-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide
[α]D 21: -7.2° (c = 1.1, EtOH) .
1H-NMR (400 MHz, CDCl3) δ: 7.88-7.79 (m, 6H), 7.67-7.58 (m, 2H), 7.50 (s, 1H), 7.36 (d, 1H, J = 7.5 Hz), 3.79-3.65 (m, 4H), 3.05 (s, 3H), 2.08 (s, 3H).
HR-MS (ESI) calcd for C22H22F3N2O4S [M+H]+, required m/z: 467.1252, found: 467.1257.
Retention time: 4.5 min.
……………………………………………….
WO 2014168103
Step B: pyrrole derivative compounds (A ‘)
[Of 16]

(Example 1) 2-bromo-1- [2- (trifluoromethyl) phenyl] propan-1-one
[Of 19]

1- [2- (trifluoromethyl) phenyl] propan-1-one 75 g (370 mmol) in t- butyl methyl ether (750 mL), and I was added bromine 1.18 g (7.4 mmol). After confirming that the stirred bromine color about 30 minutes at 15 ~ 30 ℃ disappears, cooled to 0 ~ 5 ℃, was stirred with bromine 59.13 g (370 mmol) while keeping the 0 ~ 10 ℃. After stirring for about 2.5 hours, was added while maintaining 10 w / v% aqueous potassium carbonate solution (300 mL) to 0 ~ 25 ℃, was further added sodium sulfite (7.5 g), was heated to 20 ~ 30 ℃. The solution was separated, washed in the resulting organic layer was added water (225 mL), to give t- butyl methyl ether solution of the title compound and the organic layer was concentrated under reduced pressure (225 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.91 (3H, D, J = 4.0 Hz), 4.97 (1H, Q, J = 6.7 Hz), 7.60 ~ 7.74 (4H, M).
(Example 2) 2-cyano-3-methyl-4-oxo-4- [2- (trifluoromethyl) phenyl] butanoate
[Of 20]

2-bromo-1- [2- (trifluoromethyl) phenyl] propan-1 / t- butyl methyl ether solution (220 mL) in dimethylacetamide (367 mL), ethyl cyanoacetate obtained in Example 1 53.39 g (472 mmol), potassium carbonate 60.26 g (436 mmol) were sequentially added, and the mixture was stirred and heated to 45 ~ 55 ℃. After stirring for about 2 hours, 20 is cooled to ~ 30 ℃, water (734 mL) and then extracted by addition of toluene (367 mL), washed by adding water (513 mL) was carried out in the organic layer (2 times implementation). The resulting organic layer was concentrated under reduced pressure to obtain a toluene solution of the title compound (220 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.33 ~ 1.38 (6H, M), 3.80 ~ 3.93 (2H, M), 4.28 ~ 4.33 (2H, M), 7.58 ~ 7.79 (4H, M).
(Example 3) 2-chloro-4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl
[Of 21]

The 20 ~ 30 ℃ 2-cyano-3-methyl-4-oxo-4 was obtained [2- (trifluoromethyl) phenyl] butanoate in toluene (217 mL) by the method of Example 2 ethyl acetate (362 mL) Te, after the addition of thionyl chloride 42.59 g (358 mmol), cooled to -10 ~ 5 ℃, was blown hydrochloric acid gas 52.21 g (1432 mmol), further concentrated sulfuric acid 17.83 g (179 mmol) was added, and the mixture was stirred with hot 15 ~ 30 ℃. After stirring for about 20 hours, added ethyl acetate (1086 mL), warmed to 30 ~ 40 ℃, after the addition of water (362 mL), and the layers were separated. after it separated organic layer water (362 mL) was added for liquid separation, and further 5w / v% was added for liquid separation aqueous sodium hydrogen carbonate solution (362 mL).
Subsequently the organic layer was concentrated under reduced pressure, the mixture was concentrated under reduced pressure further added toluene (579 mL), was added toluene (72 mL), and cooled to 0 ~ 5 ℃. After stirring for about 2 hours, the precipitated crystals were filtered, and washed the crystals with toluene which was cooled to 0 ~ 5 ℃ (217 mL). The resulting wet goods crystals were dried under reduced pressure at 40 ℃, the title compound was obtained (97.55 g, 82.1% yield).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.38 (3H, t, J = 7.1 Hz), 2.11 (3H, s), 4.32 (2H, Q, J = 7.1 Hz), 7.39 (1H, D, J = 7.3 Hz), 7.50 ~ 7.62 (2H, m), 7.77 (1H, d, J = 8.0 Hz), 8.31 (1H, br).
(Example 4) 4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl
[Of 22]

Example obtained by the production method of the three 2-chloro-4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylate 97.32 g (293 mmol) in ethanol (662 mL), tetrahydrofuran (117 mL), water (49 mL), sodium formate 25.91 g (381 mmol) and 5% palladium – carbon catalyst (water content 52.1%, 10.16 g) was added at room temperature, heated to 55 ~ 65 ℃ the mixture was stirred. After stirring for about 1 hour, cooled to 40 ℃ less, tetrahydrofuran (97 mL) and filter aid (KC- flock, Nippon Paper Industries) 4.87 g was added, the catalyst was filtered and the residue using ethanol (389 mL) was washed. The combined ethanol solution was used for washing the filtrate after concentration under reduced pressure, and with the addition of water (778 mL) was stirred for 0.5 hours at 20 ~ 30 ℃. The precipitated crystals were filtered, and washed the crystals with ethanol / water = 7/8 solution was mixed with (292 mL). The resulting wet goods crystals were dried under reduced pressure at 40 ℃, the title compound was obtained (86.23 g, 98.9% yield).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.35 (3H, t, J = 7.1 Hz), 2.18 (3H, s), 4.29 (2H, M), 7.40 ~ 7.61 (4H, M), 7.77 (1H, d, J = 7.9 Hz), 8.39 (1H, br).
(Example 5) (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl
[Of 23]

N to the fourth embodiment of the manufacturing method by the resulting 4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylate 65.15 g (219 mmol), N- dimethylacetamide ( 261 mL), ethylene carbonate 28.95 g (328.7 mmol), 4- dimethylaminopyridine 2.68 g (21.9 mmol) were sequentially added at room temperature, and heated to 105 ~ 120 ℃, and the mixture was stirred. After stirring for about 10 hours, toluene was cooled to 20 ~ 30 ℃ (1303 mL), and the organic layer was extracted by adding water (326 mL). Subsequently, was washed by adding water (326 mL) to the organic layer (three times). The resulting organic layer was concentrated under reduced pressure, ethanol (652 mL) was added, and was further concentrated under reduced pressure, ethanol (130 mL) was added to obtain an ethanol solution of the title compound (326 mL).
1 H NMR (400 MHz, CDCl 3 ) delta: 1.35 (3H, t, J = 7.1 Hz), 1.84 (1H, Broad singlet), 2.00 (3H, s), 3.63 ~ 3.77 (4H, M), 4.27 (2H , m), 7.35 ~ 7.79 (5H, m).
(Example 6) (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid
[Of 24]

Obtained by the method of Example 5 (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid ethyl / ethanol (321 mL) solution in water (128.6 mL), was added at room temperature sodium hydroxide 21.4 g (519 mmol), and stirred with heating to 65 ~ 78 ℃. After stirring for about 6 hours, cooled to 20 ~ 30 ℃, after the addition of water (193 mL), and was adjusted to pH 5.5 ~ 6.5, while maintaining the 20 ~ 30 ℃ using 6 N hydrochloric acid. was added as seed crystals to the pH adjustment by a liquid (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 6.4 mg , even I was added to water (193mL). Then cooled to 0 ~ 5 ℃, again, adjusted to pH 3 ~ 4 with concentrated hydrochloric acid and stirred for about 1 hour. Then, filtered crystals are precipitated, and washed the crystals with 20% ethanol water is cooled to 0 ~ 5 ℃ (93 mL). The resulting wet product crystals were dried under reduced pressure at 40 ℃, to give the title compound (64.32 g, 95.0% yield). 1 H NMR (400 MHz, DMSO-D 6 ) delta: 1.87 (3H, s), 3.38 ~ 3.68 (4H, M), 7.43 ~ 7.89 (5H, M).
(Example 7)
(S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
(7-1) (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
obtained by the method of Example 6 the (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 50.00 g (160 mmol), N, N- dimethylacetamide (25 mL), ethyl acetate (85 mL) was added and dissolved at room temperature (solution 1).
(7-1) (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid quinine salt
obtained by the method of Example 6 the (RS) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid 50.00 g (160 mmol), N, N- dimethylacetamide (25 mL), ethyl acetate (85 mL) was added and dissolved at room temperature (solution 1).
Quinine 31.05 g (96 mmol) in N, N- dimethylacetamide (25 mL), ethyl acetate (350 mL), was heated in water (15 mL) 65 ~ 70 ℃ was added, was added dropwise a solution 1. After about 1 hour stirring the mixture at 65 ~ 70 ℃, and slowly cooled to 0 ~ 5 ℃ (cooling rate standard: about 0.3 ℃ / min), and stirred at that temperature for about 0.5 hours. The crystals were filtered, 5 ℃ using ethyl acetate (100 mL) which was cooled to below are washed crystals, the resulting wet product crystals was obtained and dried under reduced pressure to give the title compound 43.66 g at 40 ℃ (Yield 42.9%). Furthermore, the diastereomeric excess of the obtained salt was 98.3% de. 1 H NMR (400 MHz, DMSO-D 6 ) delta: 1.30 ~ 2.20 (10H, M), 2.41 ~ 2.49 (2H, M), 2.85 ~ 3.49 (6H, M), 3.65 ~ 3.66 (1H, M), 3.88 (3H, s), 4.82 (1H, broad singlet), 4.92 ~ 5.00 (2H, m), 5.23 ~ 5.25 (1H, m), 5.60 (1H, br), 5.80 ~ 6.00 (1H, m), 7.36 ~ 7.92 (9H, M), 8.67 (1H, D, J = 4.6 Hz) (7-2) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3 diastereomeric excess of the carboxylic acid quinine salt HPLC measurements (% de) that the title compound of about 10 mg was collected, and the 10 mL was diluted with 50v / v% aqueous acetonitrile me was used as a sample solution.
Column: DAICEL CHIRALPAK IC-3 (4.6 mmI.D. × 250 mm, 3 μm)
mobile phase A: 0.02mol / L phosphorus vinegar buffer solution (pH 3)
mobile phase B: acetonitrile
solution sending of mobile phase: mobile phase A and I indicates the mixing ratio of mobile phase B in Table 1 below.
mobile phase A: 0.02mol / L phosphorus vinegar buffer solution (pH 3)
mobile phase B: acetonitrile
solution sending of mobile phase: mobile phase A and I indicates the mixing ratio of mobile phase B in Table 1 below.
[Table 1]

Detection: UV 237 nm
flow rate: about 0.8 mL / min
column temperature: 30 ℃ constant temperature in the vicinity of
measuring time: about 20 min
Injection volume: 5 μL
diastereomeric excess (% de), the title compound (retention time about 12 min), was calculated by the following equation using a peak area ratio of R-isomer (retention time of about 13 min).
% De = {[(the title compound (S body) peak area ratio) – (R body peak area ratio)] ÷ [(the title compound (S body) peak area ratio) + (R body peak area ratio)]} × 100
flow rate: about 0.8 mL / min
column temperature: 30 ℃ constant temperature in the vicinity of
measuring time: about 20 min
Injection volume: 5 μL
diastereomeric excess (% de), the title compound (retention time about 12 min), was calculated by the following equation using a peak area ratio of R-isomer (retention time of about 13 min).
% De = {[(the title compound (S body) peak area ratio) – (R body peak area ratio)] ÷ [(the title compound (S body) peak area ratio) + (R body peak area ratio)]} × 100
(Example 8)
(S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxamide (Compound (A))
(8-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3 – carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 40.00 g (63 mmol) in ethyl acetate (400 mL), was added 2N aqueous hydrochloric acid (100 mL) was stirred at room temperature and separated . The resulting organic layer was concentrated under reduced pressure (120 mL), and added ethyl acetate (200 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (120 mL).
(8-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3 – carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 40.00 g (63 mmol) in ethyl acetate (400 mL), was added 2N aqueous hydrochloric acid (100 mL) was stirred at room temperature and separated . The resulting organic layer was concentrated under reduced pressure (120 mL), and added ethyl acetate (200 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (120 mL).
(8-2) N – {[4- (methylsulfonyl) phenyl] amino} oxamic acid 2 – ((S) -3- methyl-4 – {[4- (methylsulfonyl) phenyl] carbamoyl} -2- [ 2- (trifluoromethyl) phenyl] -1H- pyrrol-1-yl) ethyl
ethyl acetate (240 mL), was mixed tetrahydrofuran (80 mL) and oxalyl chloride 20.72 g (163 mmol), and cooled to 10 ~ 15 ℃ was. Then the resulting solution was added while keeping the 10 ~ 15 ℃ Example (8-1) and stirred for about 1 hour by heating to 15 ~ 20 ℃. After stirring, acetonitrile (120 mL) and pyridine 2.46 g (31 mmol) was added and the reaction mixture was concentrated under reduced pressure (120 mL), acetonitrile (200 mL) was added and further concentrated under reduced pressure (120 mL).
ethyl acetate (240 mL), was mixed tetrahydrofuran (80 mL) and oxalyl chloride 20.72 g (163 mmol), and cooled to 10 ~ 15 ℃ was. Then the resulting solution was added while keeping the 10 ~ 15 ℃ Example (8-1) and stirred for about 1 hour by heating to 15 ~ 20 ℃. After stirring, acetonitrile (120 mL) and pyridine 2.46 g (31 mmol) was added and the reaction mixture was concentrated under reduced pressure (120 mL), acetonitrile (200 mL) was added and further concentrated under reduced pressure (120 mL).
After completion concentration under reduced pressure, acetonitrile (200 mL) was added and cooled to 10 ~ 15 ℃ (reaction 1).
Acetonitrile (240mL), pyridine 12.39 g (157 mmol), 4- were successively added (methylsulfonyl) aniline 26.85 g (157 mmol), the reaction solution 1 was added while maintaining the 10 ~ 15 ℃, the 20 ~ 25 ℃ and the mixture was stirred and heated to about 1 hour.
The resulting reaction solution in acetonitrile (40 mL), 2 N hydrochloric acid water (120 mL), was added sodium chloride (10.0 g) was stirred, and the layers were separated. Again, 2N aqueous hydrochloric acid to the organic layer (120 mL), was added sodium chloride (10.0 g) was stirred, and the layers were separated. After filtering the resulting organic layer was concentrated under reduced pressure (400 mL). Water (360 mL) was added to the concentrated liquid, after about 1 hour stirring, the crystals were filtered, washed with 50v / v% aqueous acetonitrile (120 mL), wet product of the title compound (undried product, 62.02 g) and obtained. 1 H NMR (500 MHz, DMSO-D 6 ) delta: 1.94 (s, 3H), 3.19 (s, 3H), 3.20 (s, 3H), 3.81 (t, 1H), 4.12 (t, 1H), 4.45 ( t, 2H, J = 5.81 Hz), 7.62 (t, 1H, J = 4.39 Hz), 7.74 (t, 2H, J = 3.68 Hz), 7.86 (dd, 3H), 7.92 (dd, 3H, J = 6.94 , 2.13 Hz), 7.97 (DD, 2H, J = 6.80, 1.98 Hz), 8.02 (DD, 2H), 10.03 (s, 1H), 11.19 (s, 1H)
(8-3) (S)-1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (Compound (A)) ( the resulting wet product crystals 8-2), t- butyl methyl ether (200 mL), acetonitrile (40 mL), 48w / w potassium hydroxide aqueous solution (16 g) and water (200 mL) was added, I was stirred for about 2 hours at 25 ~ 35 ℃. After stirring, and the mixture is separated, the resulting organic layer was concentrated under reduced pressure (120 mL), ethanol (240 mL) was added and further concentrated under reduced pressure (120 mL). After completion concentration under reduced pressure, ethanol (36 mL), and heated in water (12 mL) was added 35 ~ 45 ℃, while maintaining the 35 ~ 45 ℃ was added dropwise water (280 mL), and was crystallized crystals. After cooling the crystal exudates to room temperature, I was filtered crystal. Then washed with crystals 30v / v% aqueous ethanol solution (80 mL), where it was dried under reduced pressure at 40 ℃, the title compound was obtained in crystalline (26.26 g, 89.7% yield). Moreover, the enantiomers of the resulting crystals was 0.3%.
1 H NMR (400 MHz, CDCl 3 ) delta: 1.74 (1H, Broad singlet), 2.08 (3H, s), 3.04 (3H, s), 3.63 ~ 3.80 (4H, M), 7.36 (1H, D, J = 7.2 Hz), 7.48 (1H, s), 7.58 ~ 7.67 (2H, M), 7.77 ~ 7.90 (6H, M).
(8-4) (S)-1-(2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole -3- HPLC method for measuring the amount enantiomer carboxamide (%) and collected the title compound of about 10 mg is, what was the 10 mL was diluted with 50v / v% aqueous acetonitrile to obtain a sample solution.
see
(Example 12) (S) -1- (2- hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxamide (Compound (A)) Preparation of 2
(12-1) (S)-1-(2-hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H – pyrrole-3-carboxylic acid
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 10.00 g (16 mmol) in t- butyl methyl ether (90 mL), water (10 mL) 36w / w% aqueous hydrochloric acid ( 5 mL) was added and stirring at room temperature and separated. The resulting organic layer was concentrated under reduced pressure (30 mL), was added ethyl acetate (50 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (30 mL).
obtained by the method of Example 7 (S) -1- (2- hydroxyethyl) -4-methyl-5- [2- (trifluoromethyl) phenyl] -1H- pyrrole 3-carboxylic acid (8α, 9R) -6′- methoxycinnamate Conan-9-ol 10.00 g (16 mmol) in t- butyl methyl ether (90 mL), water (10 mL) 36w / w% aqueous hydrochloric acid ( 5 mL) was added and stirring at room temperature and separated. The resulting organic layer was concentrated under reduced pressure (30 mL), was added ethyl acetate (50 mL), and further concentrated under reduced pressure to obtain a solution containing the title compound (30 mL).
(12-2) N – {[4- (methylsulfonyl) phenyl] amino} oxamic acid 2 – ((S) -3- methyl-4 – {[4- (methylsulfonyl) phenyl] carbamoyl} -2- [ 2- (trifluoromethyl) phenyl] -1H- pyrrol-1-yl) ethyl
ethyl acetate (50 mL), was mixed with tetrahydrofuran (20 mL) and oxalyl chloride 5.18 g (41 mmol), and cooled to 0 ~ 5 ℃ was.Then the resulting solution was added in Examples while maintaining the 0 ~ 5 ℃ (12-1), and the mixture was stirred for 6 hours at 0 ~ 10 ℃. After stirring, acetonitrile (30 mL) and pyridine 0.62 g (8 mmol) was added and the reaction mixture was concentrated under reduced pressure (30 mL), acetonitrile (50 mL) was added, and further concentrated under reduced pressure (30 mL).
ethyl acetate (50 mL), was mixed with tetrahydrofuran (20 mL) and oxalyl chloride 5.18 g (41 mmol), and cooled to 0 ~ 5 ℃ was.Then the resulting solution was added in Examples while maintaining the 0 ~ 5 ℃ (12-1), and the mixture was stirred for 6 hours at 0 ~ 10 ℃. After stirring, acetonitrile (30 mL) and pyridine 0.62 g (8 mmol) was added and the reaction mixture was concentrated under reduced pressure (30 mL), acetonitrile (50 mL) was added, and further concentrated under reduced pressure (30 mL).
After concentration under reduced pressure end, is added acetonitrile (10 mL) and oxalyl chloride 0.10 g (1 mmol), and cooled to 0 ~ 5 ℃ (reaction 1).
Acetonitrile (30mL), pyridine 3.15 g (40 mmol), 4- were successively added (methylsulfonyl) aniline 6.71 g (39 mmol), the reaction solution 1 was added while maintaining the 10 ~ 15 ℃, the 20 ~ 25 ℃ and the mixture was stirred and heated to about 1 hour.
Insolubles from the resulting reaction solution was filtered, washed with acetonitrile (10 mL), and stirred for about 2 hours the addition of water (15 mL), followed by dropwise addition of water (75 mL) over about 1 hour . After about 1 hour stirring the suspension was filtered crystals were washed with 50v / v% aqueous acetonitrile (20 mL), wet product of the title compound (undried product, 15.78 g) to give a. 1 H NMR (500 MHz, DMSO-D 6 ) delta: 1.94 (s, 3H), 3.19 (s, 3H), 3.20 (s, 3H), 3.81 (t, 1H), 4.12 (t, 1H), 4.45 ( t, 2H, J = 5.81 Hz), 7.62 (t, 1H, J = 4.39 Hz), 7.74 (t, 2H, J = 3.68 Hz), 7.86 (dd, 3H), 7.92 (dd, 3H, J = 6.94 , 2.13 Hz), 7.97 (DD, 2H, J = 6.80, 1.98 Hz), 8.02 (DD, 2H), 10.03 (s, 1H), 11.19 (s, 1H)
(12-3) (S)-1- (2-hydroxyethyl) -4-methyl -N- [4- (methylsulfonyl) phenyl] -5- [2- (trifluoromethyl) phenyl] -1H- pyrrole-3-carboxamide (Compound (A)) ( the resulting wet product crystals 12-2), t- butyl methyl ether (50 mL), acetonitrile (10 mL), 48w / w potassium hydroxide aqueous solution (4 g) and water (50 mL) was added, 15 I was about 2 hours of stirring at ~ 25 ℃. After stirring, and the mixture is separated, the resulting organic layer was concentrated under reduced pressure (30 mL), was added ethanol (60 mL), was further concentrated under reduced pressure (30 mL). After completion concentration under reduced pressure, ethanol (14 mL), after addition of water (20 mL), was added a seed crystal, and was crystallized crystals. After dropwise over about 1 hour water (50 mL), and about 1 hour stirring, and crystals were filtered off. Then washed with crystals 30v / v% aqueous ethanol solution (10 mL), where it was dried under reduced pressure at 40 ℃, the title compound was obtained in crystal (6.36 g, 87.0% yield). Moreover, the enantiomers of the resulting crystals was 0.05%. Enantiomers amount, I was measured by the method of (Example 8-4). 1 H NMR (400 MHz, CDCl 3 ) delta: 1.74 (1H, Broad singlet), 2.08 (3H, s), 3.04 (3H, s), 3.63 ~ 3.80 (4H, M), 7.36 (1H, D, J = 7.2 Hz), 7.48 (1H, s), 7.58 ~ 7.67 (2H, m), 7.77 ~ 7.90 (6H, m).
………………………………………………
Patent literature
Patent Document 1: International Publication WO2006 / 012642 (US Publication US2008-0234270)
Patent Document 2: International Publication WO2008 / 056907 (US Publication US2010-0093826)
Patent Document 3: Pat. No. 2,082,519 JP (US Patent No. 5,616,599 JP)
Patent Document 4: Pat. No. 1,401,088 JP (US Pat. No. 4,572,909)
Patent Document 5: US Pat. No. 3,025,292
Patent Document 2: International Publication WO2008 / 056907 (US Publication US2010-0093826)
Patent Document 3: Pat. No. 2,082,519 JP (US Patent No. 5,616,599 JP)
Patent Document 4: Pat. No. 1,401,088 JP (US Pat. No. 4,572,909)
Patent Document 5: US Pat. No. 3,025,292
Angiotensin II receptor 桔抗 agent
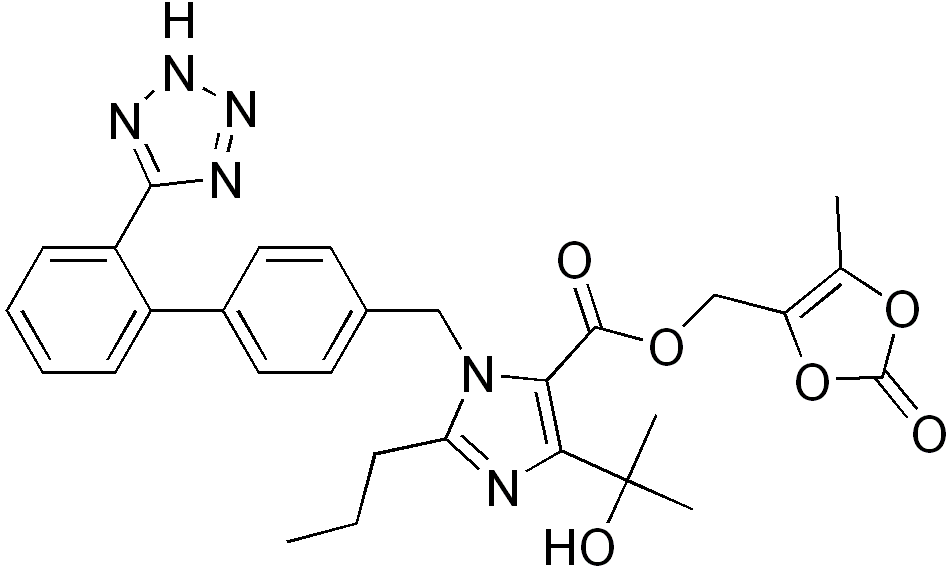
Angiotensin II receptor 桔抗 agent used as the component (A), olmesartan medoxomil, olmesartan cilexetil, losartan, candesartan cilexetil, valsartan, biphenyl tetrazole compounds such as irbesartan, biphenyl carboxylic acid compounds such as telmisartan, eprosartan, agile Sultan, and the like, preferably, a biphenyl tetrazole compound, more preferably, olmesartan medoxomil, is losartan, candesartan cilexetil, valsartan or irbesartan, particularly preferred are olmesartan medoxomil, losartan or candesartan cilexetil, Most preferably, it is olmesartan medoxomil.
Olmesartan medoxomil, JP-A-5-78328, US Patent No. 5,616,599
is described in Japanese or the like, its chemical name is (5-methyl-2-oxo-1,3-dioxolen-4-yl ) methyl 4- (1-hydroxy-1-methylethyl) -2-propyl-1 – in [2 ‘(1H- tetrazol-5-yl) biphenyl-4-ylmethyl] imidazole-5-carboxylate, Yes, olmesartan medoxomil of the present application includes its pharmacologically acceptable salt.
is described in Japanese or the like, its chemical name is (5-methyl-2-oxo-1,3-dioxolen-4-yl ) methyl 4- (1-hydroxy-1-methylethyl) -2-propyl-1 – in [2 ‘(1H- tetrazol-5-yl) biphenyl-4-ylmethyl] imidazole-5-carboxylate, Yes, olmesartan medoxomil of the present application includes its pharmacologically acceptable salt.
OLMESARTAN
Losartan (DUP-753) is, JP 63-23868, is described in US Patent No. 5,138,069 JP like, and its chemical name is 2-butyl-4-chloro-1- [2 ‘ – The (1H- tetrazol-5-yl) biphenyl-4-ylmethyl] -1H- is imidazol-5-methanol, application of losartan includes its pharmacologically acceptable salt (losartan potassium salt, etc.).
LOSARTAN
Candesartan cilexetil, JP-A-4-364171, EP-459136 JP, is described in US Patent No. 5,354,766 JP like, and its chemical name is 1- (cyclohexyloxycarbonyloxy) ethyl-2 ethoxy-1- [2 ‘one (1H- tetrazol-5-yl) -4-Bife~eniru ylmethyl] -1H- benzimidazole-7-carboxylate is a salt application of candesartan cilexetil, which is a pharmacologically acceptable encompasses.
Valsartan (CGP-48933), the JP-A-4-159718, are described in EP-433983 JP-like, and its chemical name, (S) -N- valeryl -N- [2 ‘- (1H- tetrazol – It is a 5-yl) biphenyl-4-ylmethyl) valine, valsartan of the present application includes its pharmacologically acceptable ester or a pharmacologically acceptable salt thereof.
Irbesartan (SR-47436), the Japanese Patent Publication No. Hei 4-506222, is described in JP WO91-14679 publication, etc., its chemical name, 2-N–butyl-4-spiro cyclopentane-1- [2′ The (tetrazol-5-yl) biphenyl-4-ylmethyl] -2-imidazoline-5-one, irbesartan of the present application includes its pharmacologically acceptable salts.
Eprosartan (SKB-108566) is described in US Patent No. 5,185,351 JP etc., the chemical name, 3- [1- (4-carboxyphenyl-methyl) -2-n- butyl – imidazol-5-yl] The 2-thienyl – methyl-2-propenoic acid, present in eprosartan, the carboxylic acid derivatives, pharmacologically acceptable ester or a pharmacologically acceptable salt of a carboxylic acid derivative (eprosartan mesylate, encompasses etc.).
Telmisartan (BIBR-277) is described in US Patent No. 5,591,762 JP like, and its chemical name is 4 ‘- [[4 Mechiru 6- (1-methyl-2-benzimidazolyl) -2 – is a propyl-1-benzimidazolyl] methyl] -2-biphenylcarboxylic acid, telmisartan of the present application includes its carboxylic acid derivative, a pharmacologically acceptable ester or a pharmacologically acceptable salt thereof of carboxylic acid derivatives .
Agile Sultan, is described in Patent Publication No. 05-271228 flat JP, US Patent No. 5,243,054 JP like, and its chemical name is 2-ethoxy-1 {[2 ‘- (5-oxo-4,5-dihydro 1,2,4-oxadiazole-3-yl) biphenyl-4-yl] methyl} -1H- benzo [d] imidazole-7-carboxylic acid (2-Ethoxy-1 {[2 ‘- (5- oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) biphenyl-4-yl] is a methyl} -1H-benzo [d] imidazole-7-carboxylic acid).











































.svg)

