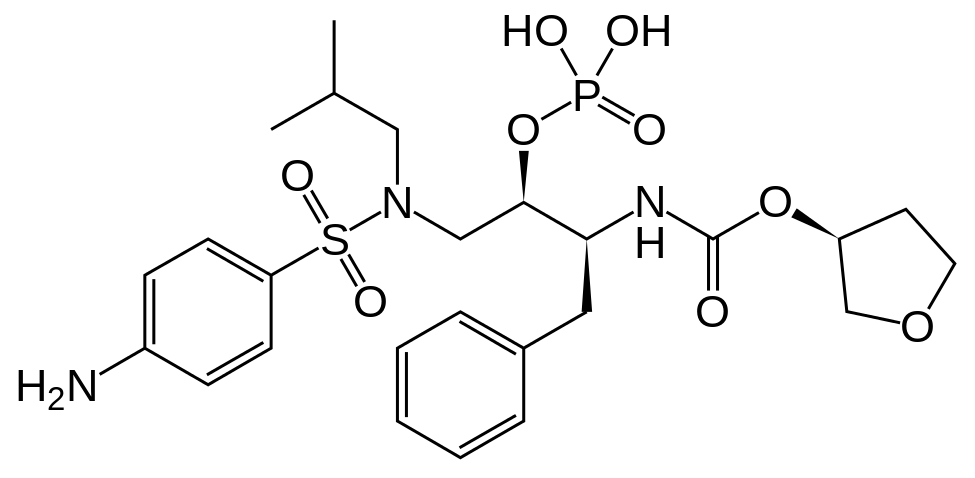
Fosamprenavir
BASE
CAS 226700-79-4
[(1S,2R)-3-[[(4-Aminophenyl)sulfonyl](2-methylpropyl)amino]-1-(phenylmethyl)-2-(phosphonooxy)propyl]carbamic acidC-[(3S)-tetrahydro-3-furanyl] ester
Additional Names: (3S)-tetrahydro-3-furyl [(aS)-a-[(1R)-1-hydroxy-2-(N1-isobutylsulfanilamido)ethyl]phenethyl]carbamate dihydrogen phosphate (ester)
Manufacturers’ Codes: VX-175
Molecular Formula: C25H36N3O9PS
Molecular Weight: 585.61
Percent Composition: C 51.27%, H 6.20%, N 7.18%, O 24.59%, P 5.29%, S 5.48%
WO 9933815 PRODUCT PATENT
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| {[(2R,3S)-1-[N-(2-methylpropyl)(4-aminobenzene)sulfonamido]-3-({[(3S)-oxolan-3-yloxy]carbonyl}amino)-4-phenylbutan-2-yl]oxy}phosphonic acid | |
| CLINICAL DATA | |
| TRADE NAMES | Lexiva |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a604012 |
| |
| |
| Oral | |
| PHARMACOKINETIC DATA | |
| BIOAVAILABILITY | Unknown |
| PROTEIN BINDING | 90% |
| METABOLISM | Hydrolysed to amprenavirand phosphate in GI tractepithelium |
| HALF-LIFE | 7.7 hours |
| EXCRETION | Fecal (as metabolites of amprenavir) |
| IDENTIFIERS | |
| 226700-81-8 | |
| J05AE07 | |
| PUBCHEM | CID 131536 |
| DRUGBANK | DB01319 |
| CHEMSPIDER | 116245 |
| UNII | WOU1621EEG |
| CHEMBL | CHEMBL1664 |
| NIAID CHEMDB | 082186 |
| CHEMICAL DATA | |
| FORMULA | C25H36N3O9PS |
| 585.608 g/mol 623.700 g/mol (calciumsalt) | |

Calcium salt
CAS 226700-81-8
Manufacturers’ Codes: GW-433908G
Trademarks: Lexiva (GSK); Telzir (GSK)
Molecular Formula: C25H34CaN3O9PS
Molecular Weight: 623.67
Percent Composition: C 48.15%, H 5.49%, Ca 6.43%, N 6.74%, O 23.09%, P 4.97%, S 5.14%
Properties: White microcrystalline needles, mp 282-284°. Soly in water (25°): 0.31 mg/ml.
Melting point: mp 282-284°
Therap-Cat: Antiviral.
Fosamprenavir (marketed by ViiV Healthcare as the calcium salt), under the trade names Lexiva (U.S.) and Telzir (Europe) is apro-drug of the protease inhibitor and antiretroviral drug amprenavir. The FDA approved it October 20, 2003, while the EMEA approved it on July 12, 2004. The human body metabolizes fosamprenavir in order to form amprenavir, which is the active ingredient. That metabolization increases the duration that amprenavir is available, making fosamprenavir a slow-release version of amprenavir and thus reducing the number of pills required versus standard amprenavir.
A head-to-head study with lopinavir[1] showed the two drugs to have comparable potency, but patients on fosamprenavir tended to have a higher serum cholesterol. Fosamprenavir’s main advantage over lopinavir is that it is cheaper.
PATENT
Fosamprenavir calcium has HIV aspartyl protease inhibitory activity and is particularly well suited for inhibiting HIV-1 and HIV-2 viruses; it is chemically known as calcium (3S) tetrahydro-3-furanyl(l S,2R)-3-[[(4-aminophenyl) sulfonyl] (isobutyl) amino]- l-benzyl-2- (phosphonooxy)propyl carbamate and represented by formula la.
(la)
There are very few references available in the literature for preparation of fosamprenavir and its intermediates. Patent US 5 585 397 provides process for preparation of fosamprenavir intermediate (IV), as depicted in scheme 1 , wherein it is purified using silica gel chromatography, however it does not provide any purity data. Purification by column chromatography is not suitable on commercial scale, since it is time consuming, requires large volume of solvents and is very much laborious.

Scheme 1: Process for preparation of fosamprenavir intermediate (IV) as given in US 5
585 397 Another patent US 6 281 367, provides process for preparation of fosamprenavir intermediate (IV) as depicted in scheme 2, but it does not provide any method for purification of compound (IV).

P= amine protecting
group deprotection

Scheme 2: Process for preparation of fosamprenavir intermediate (VI) as given in US 6
281 367
The patent US 6 514 953 provides process for preparation of fosamprenvair calcium (la) utilizing compound (IV), as depicted in Scheme 3, however it does not provide purity of fosamprenavir calcium (la) or the intermediates thereof.

Aq. soln. of Ca(OAc)2
monohydrate

(la) crude (la)
Scheme 3: Process for preparation of fosamprenavir Calcium (la) as given in US 6 514
953 Another patent, US 6 436 989, which is product patent for fosamprenavir salts, provide process for preparation of fosamprenavir sodium salt (VII) from compound (IV) as depicted in Scheme 4:

(VIA)
(V)
3 eq. NaHC03
resin column,
lyophilize

Scheme 4: Process for preparation of fosamprenavir sodium (VII) as given in US 6 436989. US 6 436 989 provides compound (V) and (VIA) with an HPLC purity of 90% and 92% respectively, however purity of fosamprenavir sodium salt (VII) is not mentioned. This patent provides fosmaprenavir salt intermediates with very low HPLC purity. The prior art literature describes synthesis of fosamprenavir calcium and its intermediates and like any synthetic compound, fosmaprenavir calcium can contain number of impurities from various source like starting material, reaction by-products, degradation, isomeric impurities etc. The prior art documents for fosamprenavir calcium does not provide any information for the impurities that may have been formed from the various synthetic processes provided therein.
Fosamprenavir calcium i.e. calcium (3S) tetrahydro-3-furanyl(lS,2R)-3-[[(4-aminophenyl) sulfonyl] (isobutyl) amino]- 1 -benzyl-2-(phosphonooxy)propyl carbamate (la), is a chiral substrate containing three asymmetrical carbon centre resulting into eight stereoisomers.
Different isomers of a chiral drug molecule bind differently to target receptors, one isomer of a drug may have a desired beneficial effect while the other may cause serious and undesired side effects or sometimes even beneficial but entirely different effects, hence in the drug molecules the effective isomer is preferred in pure form, free of other undesired isomers, thus fosamprenavir calcium free of its other stereoisomer would always be preferred.
The methods described above for preparation of fosamprenavir does not describe suitable methods to minimize formation of R-isomer impurity (lb)

(lb)
One of the approach to minimize R-isomer impurity (lb) is to use highly pure intermediate (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate (Ila), in the synthesis of fosamprenavir. US 5 585 397 provides process for preparation of N-succinimidlyl-(S)-3-tetrahydrofuryl carbonate (Ila), however it does not provide any method for purification neither does it provide any purity data for the same. The PCT application WO 94/18192 provides process for preparation (S)-3-tetrahydrofuranyl- N-succinimidyl carbonate (Ila) as depicted in scheme 5. The application discloses recrystallization of compound (Ila) from EtOAc/hexane. At our hands, crystallization of compound (Ila) from ethyl acetate/hexane provided compound (Ila) containing the intermediate R-isomer impurity compound (lib) upto 0.37% area percentage of HPLC, which is not suitable for its use in the synthesis of fosamprenavir substantially free of R-isomer impurity (lb).

(VIII) (IX) (II)
a= S-isomer a= S-isomer
b= R-isomer b= R-isomer
Scheme 5: process for preparation of (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate
Commercially available (S)-3-tetrahydrofuranol (Villa) contains upto 5% area percentage of HPLC of (R)-3-tetrahydrofuranyl (Vlllb), which on reaction with N,N-disuccinimidyl carbonate (IX) results in (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate (Ila) containing upto 2.5% area percentage of HPLC of the R-isomer impurity, (R)-3-tetrahydrofuranyl-N- succinimidyl carbonate (lib). This impure (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate (Ila) when converted to fosamprenavir calcium (la) by series of reaction, results into fosamprenavir calcium containing upto 2.0 % area percentage of HPLC of (3R) tetrahydro-3- furanyl(l S,2R)-3-[[(4-aminophenyl) sulfonyl] (isobutyl) amino]- 1 -benzy 1-2- (phosphonooxy)propyl carbamate (lb), which is undesired isomer of fosamprenavir calcium. Impurities of any form are undesirable in the active pharmaceutical product since it may have adverse effect on the patient to be treated.
The purity of API produced is clearly a necessary condition for commercialization. The impurities produced in the manufacturing process must be limited to very small amount and are preferred to be substantially absent. The ICH Q7A guidance for API manufacturers requires that process impurities must be maintained below set limits utilizing various parameters. In the United States the Food and Drug Administration guidelines, would mostly limit the amount of impurities present in the API, similarly in other countries the impurity levels would be defined in their respective pharmacopeias.
The process for preparation of fosamprenavir calcium (la) of present invention is as depicted in scheme 5.

crude fosamprenavir calcium (la)
Example 2: Preparation of pure fosamprenavir calcium (I).
Mixture of 100 g (0.23 mol) (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4- nitrobenzene sulphonamide (III), 65 g (0.28 mol) (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate (Ila) (of Example 1) and 24 g (0.23) triethylamine in 800 ml dichloromethane was stirred at ambient temperature for 4 hours, extracted with 10% sodium bicarbonate solution. The organic layer was separated, washed with water and concentrated. To the concentrated mass was added 1000 ml methanol and heated to 60-65°, cooled to 25°C and solid was filtered, washed with methanol and dried. Mixture of 100 g (0.186 mol) (3S)-tetrahydro-3-furyl N-[(l S,2R)-l-benzyl-2-hydroxy-3-(N- isobutyl-4-nitrobenzene sulphonamido) propyl] carbamate (IV) and 200 ml pyridine was cooled to 0-10°C and 70.0 g (0.456 mol) of POCl3 was added and stirred at ambient temperature for 4 hours, 400 ml methyl isobutyl ketone was added, cooled and 1 : 1 cone. HC1- water was added. Mixture was heated to 50°C for 1 hour, cooled to 25-30°C. Organic layer was separated, washed with water and partially concentrated; 500 ml water and 31.5 g sodium bicarbonate was added and stirred. The organic layer was separated and 100 ml ethylacetate, 400 ml methanol and 5.0 g Pd/C was added. The reaction mass was stirred under hydrogen pressure for 4 hours at 30°C. The mixture was filtered, catalyst washed with methanol. The filtrate was heated to 50°C and 33.0 g (0.186 mol) calcium acetate monohydrate in 100 ml water was added and stirred for 30 minutes. Cooled to 30°C and stirred. Solid was filtered, washed with 1 : 1 mixture of methanol-water and dried to obtain crude fosamprenavir calcium. 65 g (0.104 mol) crude fosamprenavir calcium and 1 170 ml denatured ethanol was heated to 70-72°C, charcaolized. Water (138 ml) was added and mixture stirred for 30 minutes. Cooled to ambient temperature and stirred. Solid filtered, washed with 1 : 1 ethanol-water and dried. Methanol (315 ml) was added to the solid, stirred and filtered. The filtrate was concentrated under vacuum to obtain solid, which was dried to obtain 37.5 g pure fosamprenavir calcium. HPLC purity: fosamprenavir calcium (la): 99.85%; R-isomer impurity (lb): 0.05%; all other individual impurities less than 0.1%.

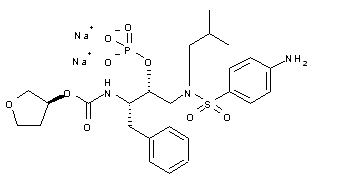
Fosamprenavir sodium, GW-433908A, 908, VX-175(free acid)
………………………………….
PAPER
Org. Biomol. Chem., 2004,2, 2061-2070
DOI: 10.1039/B404071F
Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors(Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.

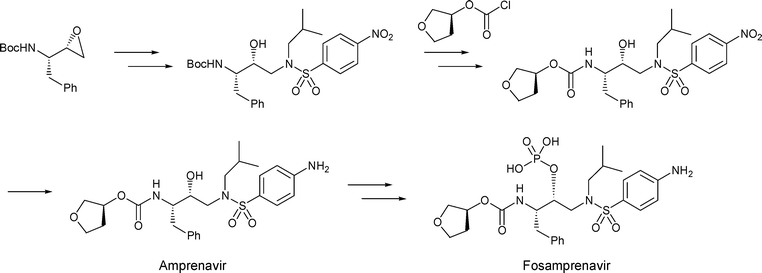
…………………
EP 0659181; EP 0885887; JP 1996501299; US 5585397; WO 9405639
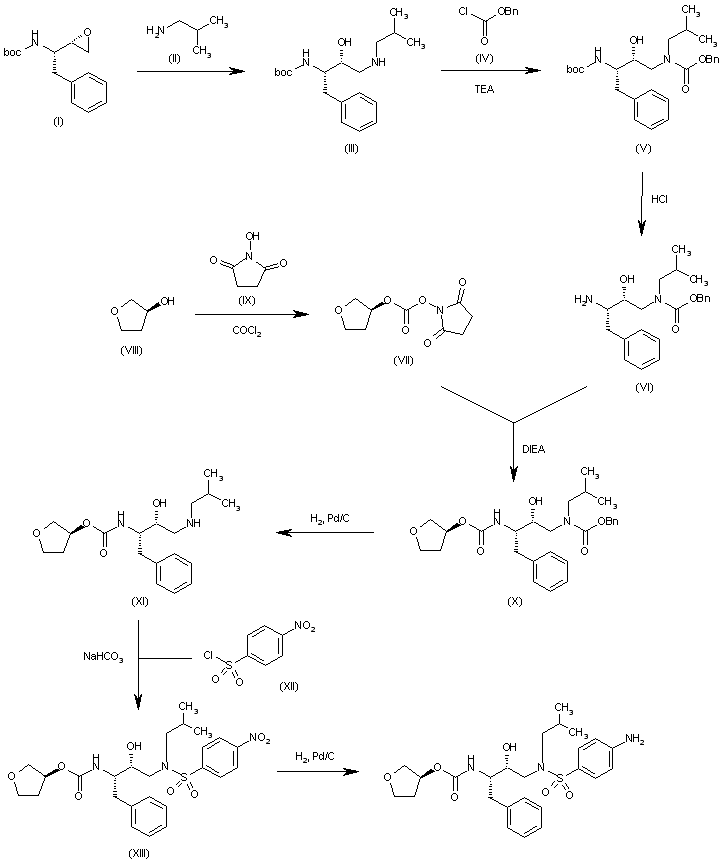
The reaction of the chiral epoxide (I) with isobutylamine (II) in refluxing ethanol gives the secondary amine (III), which is protected with benzyl chloroformate (IV) and TEA, yielding the dicarbamate (V). Selective deprotection of (V) with dry HCl in ethyl acetate affords the primary amine (VI), which is treated with 3(S)-tetrahydrofuryl N-succinimidinyl carbonate (VII) (prepared by condensation of tetrahydrofuran-3(S)-ol (VIII) with phosgene and N-hydroxysuccinimide (IX)) and DIEA in acetonitrile to provide the corresponding carbamate (X). The deprotection of (X) by hydrogenation with H2 over Pd/C in ethanol gives the secondary amine (XI), which is condensed with 4-nitrophenylsulfonyl chloride (XII) by means of NaHCO3 in dichloromethane/water to yield the sulfonamide (XIII). Finally, the nitro group of (XIII) is reduced with H2 over Pd/C in ethyl acetate to afford the target
………………………….
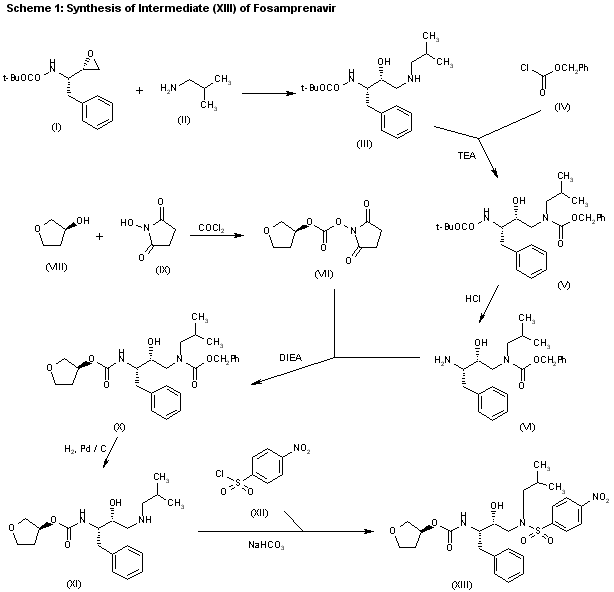
The reaction of the chiral epoxide (I) with isobutylamine (II) in refluxing ethanol gives the secondary amine (III), which is protected with benzyl chloroformate (IV) and TEA, yielding dicarbamate (V). Selective deprotection of (V) with dry HCl in ethyl acetate affords the primary amine (VI), which is treated with 3(S)-tetrahydrofuryl N-succinimidinyl carbonate (VII) — obtained by reaction of tetrahydrofuran-3(S)-ol (VIII) first with phosgene and then with N-hydroxysuccinimide (IX) — and DIEA in acetonitrile to provide the corresponding carbamate (X). Deprotection of (X) by hydrogenation with H2 over Pd/C in ethanol gives the secondary amine (XI), which is condensed with 4-nitrophenylsulfonyl chloride (XII) by means of NaHCO3 in dichloromethane/water to yield the sulfonamide intermediate (XIII).
……………………………
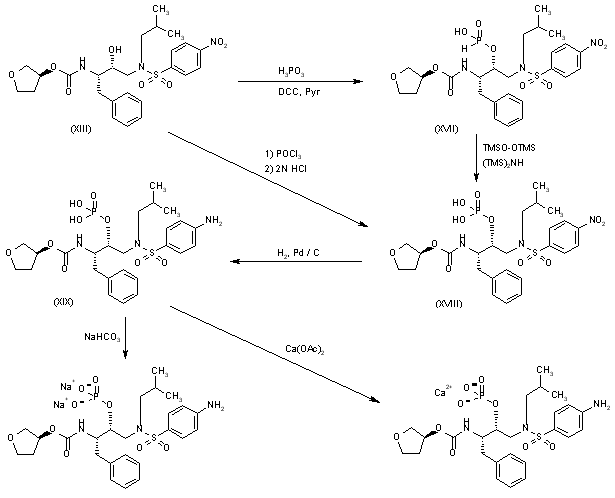
Esterification of the OH group of compound (XIII) with PO3H3 by means of DCC in hot pyridine gives the corresponding phosphite (XVII), which is oxidized with bis(trimethylsilyl)peroxide in bis(trimethylsilyl)azane to yield the expected phosphate (XVIII). Reduction of the nitro group of (XVIII) with H2 over Pd/C in ethyl acetate affords fosamprenavir (XIX). Finally, fosamprenavir (XIX) is treated with aqueous NaHCO3 or with calcium acetate in water to provide the corresponding salts. Alternatively, the phosphate (XIX) can be obtained directly by reaction of intermediate (XIII) with POCl3 in pyridine, followed by hydrolysis with 2N HCl.
………………………………………..
HIV protease inhibitor; water soluble prodrug of amprenavir, q.v. Prepn: R. D. Tung et al., WO 9933815;eidem, US 6559137 (1999, 2003 both to Vertex).
Prepn of crystalline calcium salt: I. G. Armitage et al., WO 0004033 (2000 to Glaxo); eidem, US 6514953 (2003 to SKB).
Clinical pharmacokinetics: C. Falcoz et al., J. Clin. Pharmacol. 42, 887 (2002).
Review of pharmacology and clinical experience in HIV: T. M. Chapman et al.,Drugs 64, 2101-2124 (2004); C. Arvieux, O. Tribut,ibid. 65, 633-659 (2005).
References
- Eron J Jr, Yeni P, Gathe J Jr et al. (2006). “The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trial”. Lancet 368 (9534): 476–82.doi:10.1016/S0140-6736(06)69155-1. PMID 16890834.
| WO1994005639A1* | Sep 7, 1993 | Mar 17, 1994 | Vertex Pharma | Sulfonamide inhibitors of hiv-aspartyl protease |
| WO1994018192A1 | Feb 7, 1994 | Aug 18, 1994 | Merck & Co Inc | Piperazine derivatives as hiv protease inhibitors |
| INKO02772010A | Title not available | |||
| US5585397 | Sep 7, 1993 | Dec 17, 1996 | Vertex Pharmaceuticals, Incorporated | Viricides |
| US6281367 | Mar 18, 1999 | Aug 28, 2001 | Glaxo Wellcome Inc. | Process for the synthesis of HIV protease inhibitors |
| US6436989 | Dec 24, 1997 | Aug 20, 2002 | Vertex Pharmaceuticals, Incorporated | Prodrugs of aspartyl protease inhibitors |
| US6514953 | Jul 15, 1999 | Feb 4, 2003 | Smithkline Beecham Corporation | Calcium (3S) tetrahydro-3-furanyl(1S,2R)-3-[[(4-aminophenyl)sulfonyl](isobutyl)amino]-1-benzyl-2-(phosphonooxy)propylcarbamate |
| REFERENCE | ||
|---|---|---|
| 1 | * | EKHATO I VICTOR ET AL: “Isotope labeled ‘HEA/HEE’ moiety in the synthesis of labeled HIV-protease inhibitors. Part II“, JOURNAL OF LABELLED COMPOUNDS AND RADIOPHARMACEUTICALS, JOHN WILEY, CHICHESTER, GB, vol. 48, no. 3, 1 January 2005 (2005-01-01), pages 179-193, XP009112607, ISSN: 0362-4803 |
| 2 | * | MOON KIM B ET AL: “SYNTHESIS OF A CHIRAL AZIRIDINE DERIVATIVE AS A VERSATILE INTERMEDIATE FOR HIV PROTEASE INHIBITORS“, ORGANIC LETTERS, AMERICAN CHEMICAL SOCIETY, US, vol. 3, no. 15, 1 January 2001 (2001-01-01), pages 2349-2351, XP001179485, ISSN: 1523-7060, DOI: 10.1021/OL016147S |
| 3 | * | SORBERA, L. A. ET AL.: “FOSAMPRENAVIR“, DRUGS OF THE FUTURE, PROUS SCIENCE, ES, vol. 26, no. 3, 1 March 2001 (2001-03-01), pages 224-231, XP009001334, ISSN: 0377-8282, DOI: 10.1358/DOF.2001.026.03.615590 |

Vertex Pharmaceuticals’ Boston Campus, United States of America











 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR




