GOSOGLIPTIN
CAS 869490-23-3 FREE BASE
DIHYDROCHLORIDE..869490-47-1
GOSOGLIPTIN; UNII-GI718UO477; PF-00734200; PF-734200;
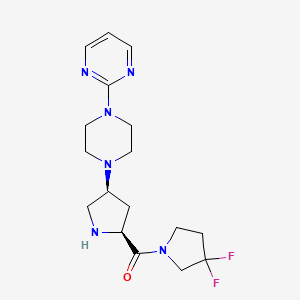
(3,3-difluoropyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-ylpiperazin-1-yl)pyrrolidin-2-yl]methanone
| Molecular Formula: | C17H24F2N6O |
|---|---|
| Molecular Weight: | 366.408866 g/mol |
| Company | Pfizer Inc. |
| Description | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Molecular Target | Dipeptidyl peptidase-4 (DPP-4) (CD26) |
| Mechanism of Action | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Latest Stage of Development | Phase II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
In vivo administration of DPP-4 inhibitors to human subjects results in higher circulating concentrations of endogenous GLP-1 and subsequent decrease in plasma glucose. Long term treatment with a DPP-4 inhibitor leads to a reduction in circulating HbA1c (glycosylated hemoglobin). DPP-4 inhibition also offers the potential to improve the insulin producing function of the pancreas through either β-cell preservation or regeneration. Therefore, DPP-4 inhibition has emerged as a promising new treatment of Type 2 diabetes
PF-734200 is a potent, selective, orally active dipeptidyl peptidase IV inhibitor. It had been in phase II clinical development at Pfizer for the treatment of type 2 diabetes; however, in 2010 the company discontinued these trials. In 2012, the product was licensed to SatRx, a spin-off of the ChemRar High Tech Center, by Pfizer on an exclusive worldwide basis (with the exception of China) for the development and commercialization as monotherapy or in combination with other therapies for the treatment of type 2 diabetes. SatRx is conducting phase II clinical trials for the treatment of type 2 diabetes.
……………………….
PAPER
New synthetic route to a dipeptidyl peptidase-4 inhibitor
Org Process Res Dev 2012, 16(3): 409
http://pubs.acs.org/doi/abs/10.1021/op200309z

(3,3-Difluoropyrrolidin-1-yl)-(2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)methanone.FREE BASE
Mp 149 °C (decomp).
[α]d = −31.1 (T = 24 °C, c = 1, CHCl3). Specific rotation of product 4 prepared using the initial route: [α]d = −31.5 (T = 24 °C, c = 1, CHCl3).
1H NMR (400 MHz; CDCl3) δ 8.30 (d, J = 4 Hz, 2H), 6.48 (t, J = 4 Hz, 1H), 3.95–3.6 (m, 9H), 3.25–2.85 (m, 4H), 2.6–2.25 (m, 7H), 1.75–1.6 (m, 1H).
13C NMR (100 MHz; CDCl3) δ 172.28; 161.55; 157.70; 127.22 (t, 1J C–F = 248 Hz), 126.22 (t, 1J C–F = 246 Hz), 109.95; 66.54; 58.87; 57.99; 52.71 (t, 2 J C–F = 32 Hz); 52.00; 50.41; 43.03; 34.46, 34.37, 34.25; 19F NMR (377 MHz, CDCl3) δ −102.1 (m, 2F).
IR (neat): 2951w, 2864w, 2799w, 2759w, 1630s, 1585vs, 1547m, 1449m, 1172m, 1254m, 1129m, 982w, 923m, 796m, 638w.
HRMS (ES, N2) Calcd for C17H24F2N6O: 367.20524, found: 367.20592.
……………………….
PAPER
(3,3-difluoro-pyrrolidin-1-yl)-((2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl)-methanone: A potent, selective, orally active dipeptidyl peptidase IV inhibitor
Bioorg Med Chem Lett 2009, 19(7): 1991
http://www.sciencedirect.com/science/article/pii/S0960894X09001966?np=y
- Pfizer Global Research & Development, Groton/New London Laboratories, Pfizer Inc, Groton, CT 06340, United States
A series of 4-substituted proline amides was evaluated as inhibitors
of dipeptidyl pepdidase IV for the treatment of type 2 diabetes.
(3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone (5) emerged as a potent (IC50 = 13 nM) and selective compound, with high oral bioavailability in preclinical species.

………………….
PATENT
WO 2005116014
http://www.google.co.in/patents/WO2005116014A1?cl=en
Example 113 (3.3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

(S)-4-Oxo-pyrrolidine-1 ,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0°C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0°C to 5°C for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0°C to 5°C. The reaction mixture turned back to aslurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0°C to 5°C.
3,3-Difluoro- pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at – 10°C to 0°C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at -10°C to 0°C. Upon completion of triethylamine addition, the mixture was stirred for 1h at 0 to 5°C. The reaction was complete by HPLC assay (-1% starting material). The reaction was quenched with water (10 volumes) at 0°C to 5 °C. The mixture was heated to 20°C to 25 °C.
The layers were separated, and the organic layer was washed with 0.5 M HCI (5 volumes). The organic layer was again washed with combined 5% NaHC03 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to -6 volumes. The mixture was held at 60°C to 65 °C for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20°C to 25 °C for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25°C to 35°C for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%.
The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65°C to 70°C. The mixture was cooled to 60°C to 65 °C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20°C to 25 °C over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35°C to 45 °C yielded 435 grams of product. HPLC purity: 96.4%.
Step 2 – (2S.4S)-2-(3.3-Dif luoro-pyrrolidine-1 -carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidine-1 – carboxylic acid tert-butyl ester A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20°C to 25°C until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60°C to 70°C until a steady temperature of 66.9°C was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to -3°C to 7°C and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at -5°C to 15°C. The reaction mixture was heated to 20°C to 25°C and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30°C to 60°C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20°C to 25CC. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45°C to 50°C. To the slurry was added heptane (10 volumes) at 45°C to 50°C over 30 minutes. The mixture was cooled to 20°C to 25°C and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35°C to 45°C yield 5.35 kg (91.3%) of the product. Step 3 – (3.3-Dif luoro-pyrrolidin-1 -yl)-f(2S.4S)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidin-2-yll- methanone Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg,
1.0 equivalent). To the slurry was added concentrated HCI (37 wt% in water, 19.1 liters, 2 volumes) slowly at 20°C to 30°C over 4 hours. The slurry went into solution after 12 liters of HCI was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5°C to 15°C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20°C to 25°C and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution.
The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50°C to 60°C. The mixture was cooled to 20°C to 25°C over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1 :1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44°C for 12 hours.
Yield: 5.7 kg, 75.9%.
m.p. 156°C. MS m/z 367 (MH+).
FREE BASE1H NMR (400 MHz, D20): δ 8.15 (d, 2H, J = 5.0 Hz, CH of pyrimidine), 6.55 (t, 1 H, J = 4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1 H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N-CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N-CH2 of piperazine), 2.97 (dd, 1 H, J = 10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1 H, H4b of proline), 2.69 (dd, 1 H, J = 10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N-CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1 H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
………………
US 2005/0256310
http://www.google.com/patents/US20050256310

This approach begins with N-t-Boc-4-oxo-l-proline (1) that undergoes a mixed anhydride activation with pivaloyl chloride at 0 °C, followed by amidation with 3,3-difluoropyrrolidine to yield the intermediate 2. Reductive amination with 1-(2-pyrimidyl)piperazine using sodium triacetoxyborohydride in THF/AcOH provided the desired stereoisomer 3 in high yield and selectivity, the undesired diastereomer being completely removed by crystallization. Deprotection of 3 with 6 N HCl, followed by neutralization with 50% NaOH and extraction provided PF-734200 (4) in good yield.
EXAMPLE 113 (3,3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

Step 1—(S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-oxo-pyrrolidine-1-carboxylic acid tert-butyl
(S)-4-Oxo-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0° C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0° C. to 5° C. for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0° C. to 5° C. The reaction mixture turned back to a slurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0° C. to 5° C. 3,3-Difluoro-pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at −10° C. to 0° C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at −10° C. to 0° C. Upon completion of triethylamine addition, the mixture was stirred for 1 h at 0 to 5° C. The reaction was complete by HPLC assay (˜1% starting material). The reaction was quenched with water (10 volumes) at 0° C. to 5 ° C. The mixture was heated to 20° C. to 25 ° C. The layers were separated, organic layer was washed with 0.5 M HCl (5 volumes). The organic layer was again washed with combined 5% NaHCO3 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to ˜6 volumes. The mixture was held at 60° C. to 65° C. for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20° C. to 25 ° C. for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25° C. to 35° C. for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%. The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65° C. to 70° C. The mixture was cooled to 60° C. to 65° C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20° C. to 25° C. over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yielded 435 grams of product. HPLC purity: 96.4%.
Step 2—(2S,4S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester
A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20° C. to 25° C. until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60° C. to 70° C. until a steady temperature of 66.9° C. was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to −3° C. to 7° C. and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at −5° C. to 15° C. The reaction mixture was heated to 20° C. to 25° C. and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30° C. to 60° C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20° C. to 25° C. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45° C. to 50° C. To the slurry was added heptane (10 volumes) at 45° C. to 50° C. over 30 minutes. The mixture was cooled to 20° C. to 25° C. and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yield 5.35 kg (91.3%) of the product.
Step 3—(3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone
Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg, 1.0 equivalent). To the slurry was added concentrated HCl (37 wt % in water, 19.1 liters, 2 volumes) slowly at 20° C. to 30° C. over 4 hours. The slurry went into solution after 12 liters of HCl was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5° C. to 15° C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20° C. to 25° C. and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution. The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50° C. to 60° C. The mixture was cooled to 20° C. to 25° C. over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1:1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44° C. for 12 hours.
Yield: 5.7 kg, 75.9%. m.p. 156° C. MS m/z 367 (MH+).
1H NMR (400 MHz, D2O): δ 8.15 (d, 2H, J=5.0 Hz, CH of pyrimidine), 6.55 (t, 1H, J=4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N—CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N—CH2 of piperazine), 2.97 (dd, 1H, J=10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1H, H4b of proline), 2.69 (dd, 1H, J=10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N—CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
……………..
PAPER

Scheme 1.


Reagents and conditions: (a) 3,3-difluoropyrrolidine hydrochloride, EDC, HOBt, TEA, DCM, rt; (b) NaBH4,
MeOH, (c) (1) trifluoromethane-sulphonyl chloride, DIPEA, DCM; (2)
2-(1-piperazinyl)pyrimidine, DCM, −10 °C; (d) 4 N HCl in dioxane, rt;
(e) 2-(1-piperazinyl)pyrimidine, NaBH(OAc)3, AcOH, DCE; (f) R1R2NH hydrochloride, EDC, HOBt TEA, DCM, 0–rt; (g) N-heterocyclic piperazine, NaBH(OAc)3, AcOH, DCE.
……………………….| Patent | Submitted | Granted |
|---|---|---|
| Therapeutic compounds [US7291618] | 2005-11-17 | 2007-11-06 |
| (2S,4S)-4-(piperazin-1-yl)pyrrolidine-2-methanone derivatives [US7465732] | 2007-05-03 | 2008-12-16 |
| THERAPEUTIC COMPOUNDS [US2007161664] | 2007-07-12 | |
| Therapeutic compounds [US2006079498] | 2006-04-13 |
 see gliptins at………….http://drugsynthesisint.blogspot.in/p/gliptin-series.html
see gliptins at………….http://drugsynthesisint.blogspot.in/p/gliptin-series.htmlhttp://organicsynthesisinternational.blogspot.in/p/gliptin-series-22.html
see gliptins at…………http://drugsynthesisint.blogspot.in/p/gliptin-series.html