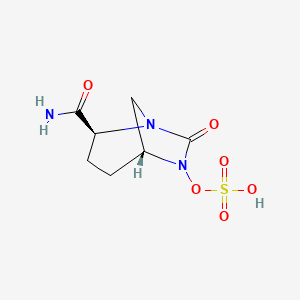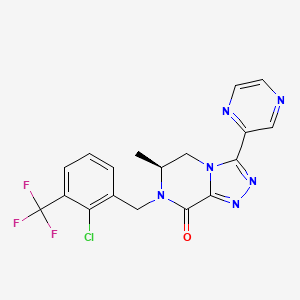
cas 1627748-32-6
1,2,4-Triazolo[4,3-a]pyrazin-8(5H)-one, 7-[[2-chloro-3-(trifluoromethyl)phenyl]methyl]-6,7-dihydro-6-methyl-3-(2-pyrazinyl)-, (6S)-
(6S)-7-[[2-chloro-3-(trifluoromethyl)phenyl]methyl]-6-methyl-3-pyrazin-2-yl-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8-one
(6S)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
Janssen Pharmaceutica Nv INNOVATOR
Michael K. Ameriks, Jason C. Rech, Brad M. Savall

(6S)-7-[[2-chloro-3-(trifluoromethyl)phenyl]methyl]-6-methyl-3-pyrazin-2-yl-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8-one
PAPER

The synthesis, SAR, and preclinical characterization of a series of substituted 6,7-dihydro[1,2,4]triazolo[4,3]pyrazin-8(5H)-one
P2X7 receptor antagonists are described. Optimized leads from this
series comprise some of the most potent human P2X7R antagonists reported
to date (IC50s < 1 nM). They also exhibit sufficient
potency and oral bioavailability in rat to enable extensive in vivo
profiling. Although many of the disclosed compounds are peripherally
restricted, compound 11d is brain
penetrant and upon oral administration demonstrated dose-dependent
target engagement in rat hippocampus as determined by ex vivo receptor
occupancy with radiotracer 5 (ED50 = 0.8 mg/kg).
Volume 26, Issue 2, 15 January 2016, Pages 257–261

Preclinical characterization of substituted 6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one P2X7 receptor antagonists
- Janssen Pharmaceutical Research & Development L.L.C., 3210 Merryfield Row, San Diego, CA 92121, United States
http://www.sciencedirect.com/science/article/pii/S0960894X15303656

Scheme 3.
PATENT
Synthesis of compounds 11d and 11l–t. Reagents and conditions: (a) Boc2O, NaOH, H2O/MeOH, 0 °C→rt (42%); (b) 2-chloro-3-trifluoromethylbenzaldehyde, Na(OAc)3BH, DCE, rt (85%); (c) methyl chlorooxoacetate, Et3N, CH2Cl2, 0 °C→rt (97%); (d) 4 N HCl/dioxane, rt, then Et3N, CH2Cl2, rt (100%); (e) Et3O+BF4−, DCM, rt, or Lawesson’s reagent, THF, 55 °C (67–99%); (f) RCONHNH2, 1-butanol, 130 °C (27–90%).
US 20140275096
http://www.google.com/patents/US20140275096
-
 Step A. tert-butyl 3-ethoxy-5,6-dihydropyrazine-1(2H)-carboxylate
Step A. tert-butyl 3-ethoxy-5,6-dihydropyrazine-1(2H)-carboxylate -
To a solution of tert-butyl 3-oxopiperazine-1-carboxylate (1 g, 5 mmol) in DCM (15 mL) was added triethyloxonium tetrafluoroborate (2.9 g, 15 mmol). Stirred for 2 h and neutralized with sat. aq NaHCO3. Layers separated and aqueous layer extracted with DCM. Combined organic layers dried over Na2SO4, filtered, and concentrated to give the title compound, which was used directly without further purification.
Step B. tert-butyl 3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate
-
To a solution of tert-butyl 3-ethoxy-5,6-dihydropyrazine-1(2H)-carboxylate (1.14 g, 5 mmol) in 1-butanol (30 mL) was added pyrazine-2-carbohydrazide (685 mg, 5 mmol). The reaction mixture was heated at reflux for 16 h. After cooling to rt, the reaction mixture was concentrated and purified by chromatography (SiO2; 2.5% MeOH in DCM) to afford the desired product as a white solid (700 mg, 50% over 2 steps). MS (ESI): mass calcd. for C14H18N6O2, 302.2; m/z found, 303.2 [M+H]+.
-
1H NMR (500 MHz, CDCl3) d 9.57 (d, J=1.4 Hz, 1H), 8.62 (d, J=2.5 Hz, 1H), 8.59-8.54 (m, 1H), 4.94 (s, 2H), 4.63-4.50 (m, 2H), 3.89 (t, J=5.4 Hz, 2H), 1.51 (s, 9H).
Step C. 3-(pyrazin-2-yl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine
-
To a solution of tert-butyl 3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate (9.3 g, 30 mmol) in DCM (100 mL) was added 1.25M HCl in EtOH (30 mL, 37.5 mmol). After 3 h, the reaction mixture was concentrated, and the resulting solid was purified by chromatography (SiO2; 10% MeOH in DCM) to provide the desired product as a white solid (3.7 g, 61%). MS (ESI): mass calcd. for C9H10N6, 202.1; m/z found, 203.1 [M+H]+. 1H NMR (400 MHz, CD3OD) δ 9.35 (d, J=1.4 Hz, 1H), 8.72 (dd, J=2.5, 1.6 Hz, 1H), 8.66 (d, J=2.6 Hz, 1H), 4.50 (t, J=5.6 Hz, 2H), 4.22 (s, 2H), 3.24 (t, J=5.6 Hz, 2H).
Step D. 2-(trimethylsilyl)ethyl 3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate
-
To a solution of 3-(pyrazin-2-yl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (1.0 g, 5.0 mmol) and N,N-diisopropylethylamine (1.7 mL, 9.9 mmol) in DMF (15 mL) was added 1-[2-(trimethylsilyl)ethoxycarbonyloxy]pyrrolidin-2,5-dione (1.5 g, 5.9 mmol). Stirred for 18 h and poured into ice cold brine (150 mL). Precipitate filtered and washed successively with water and ether to afford the desired product as a white solid (1.5 g, 89%). MS (ESI): mass calcd. for C15H22N6O2Si, 346.2; m/z found, 347.2 [M+H]+. 1H NMR (500 MHz, CDCl3) δ 9.50 (d, J=1.4 Hz, 1H), 8.56 (d, J=2.5 Hz, 1H), 8.52-8.48 (m, 1H), 4.91 (s, 2H), 4.60-4.45 (m, 2H), 4.25-4.14 (m, 2H), 3.87 (t, J=5.3 Hz, 2H), 1.07-0.92 (m, 2H), 0.01-0.04 (m, 9H).
Step E. 2-(trimethylsilyl)ethyl 8-oxo-3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate
-
To a vigorously stirred solution of 2-(trimethylsilyl)ethyl 3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate (172 mg, 0.5 mmol) in 1:1 CHCl3:MeCN (3.8 mL) was added a solution of ruthenium (IV) oxide hydrate (9.8 mg, 0.07 mmol) and sodium metaperiodate (504 mg, 2.3 mmol) in water (4.7 mL). After 4 h, the reaction mixture was diluted with water and extracted with CHCl3 (×3). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford a green oil. Purification by chromatography (SiO2; EtOAc—10% IPA/EtOAc) provided the desired product as a white solid (663 mg, 63%).
-
[0140]MS (ESI): mass calcd. for C15H20H6O3Si, 360.1; m/z found, 361.2 [M+H]+. 1H NMR (500 MHz, CDCl3) δ 9.59 (d, J=1.5 Hz, 1H), 8.63 (d, J=2.5 Hz, 1H), 8.55 (dd, J=2.5, 1.6 Hz, 1H), 4.88-4.75 (m, 2H), 4.47-4.33 (m, 2H), 4.33-4.24 (m, 2H), 1.18-1.04 (m, 2H), 0.04-(−0.02) (m, 9H).
Step F. 3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
-
To a solution of 2-(trimethylsilyl)ethyl 8-oxo-3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate (1.0 g, 2.9 mmol) in DCM (29 mL) was added TFA (5.7 mL, 75 mmol). After 1 h, the reaction mixture was concentrated. The crude residue was diluted with EtOAc, sonicated, and filtered to provide the desired product as a white solid (1.2 g, 95%). MS (ESI): mass calcd. for C9H8N6O, 216.1; m/z found, 217.1 [M+H]+. 1H NMR (500 MHz, DMSO-d6) δ 9.39 (d, J=1.1 Hz, 1H), 8.77 (q, J=2.6 Hz, 2H), 8.56 (s, 1H), 4.73-4.60 (m, 2H), 3.67-3.55 (m, 2H).
- Intermediate 1. 3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
- Intermediate 3. (±)-6-methyl-3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
-

-
Intermediate 3 was made in a manner analogous to Intermediate 1 substituting (±)-tert-butyl 2-methyl-5-oxopiperazine-1-carboxylate for tert-butyl 3-oxopiperazine-1-carboxylate in Step A. MS (ESI): mass calcd. for C10H10N6O, 230.1; m/z found, 231.1 [M+H]+.
Intermediate 4. (6S)-1-(2-chloro-3-(trifluoromethyl)benzyl)-6-methylpiperazine-2,3-dione
-
[0146]
 Step A. (S)-tert-butyl(2-aminopropyl)carbamate
Step A. (S)-tert-butyl(2-aminopropyl)carbamate -
To a solution of (S)-1,2-diaminopropane dihydrochloride (16 g, 109 mmol) in MeOH (64 mL) and water (16 mL) was added di-tert-butyl dicarbonate (28.5 g, 131 mmol) in MeOH (16 mL). The resulting solution was cooled in an ice bath, and 4N NaOH (35 mL, 140 mL) was added dropwise over 2 h. The mixture was allowed to warm to rt and stirred for a total of 20 h. The reaction was filtered, and the filtrate concentrated to remove MeOH. 200 mL EtOAc, 200 mL water, and 16 mL 1M HCl were added sequentially. The layers were separated and the aqueous layer washed with EtOAc (200 mL). The combined organic extracts were washed with 0.04M HCl (208 mL). The organic phase was separated and discarded. The aqueous phases were combined, adjusted to pH=14 with 10N NaOH (20 mL), and extracted with DCM (400 mL×2). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford the desired product as a clear oil (8.0 g, 42%). MS (ESI): mass calcd. for C8H18N2O2, 174.1; m/z found, 175.2 [M+H]+. 1H NMR (500 MHz, CDCl3) δ 5.01 (br s, 1H), 3.24-3.09 (m, 1H), 3.09-2.95 (m, 1H), 2.92-2.84 (m, 1H), 1.45 (s, 9H), 1.35-1.19 (m, 2H), 1.07 (d, J=6.4 Hz, 3H).
Step B. (6S)-tert-butyl(2-((2-chloro-3-(trifluoromethyl)benzyl)amino)propyl) carbamate
-
A solution of (S)-tert-butyl(2-aminopropyl)carbamate (4.0 g, 23 mmol) and 2-chloro-3-trifluoromethylbenzaldehyde (4.8 g, 23 mmol) in DCE (100 mL) was stirred at rt for 2 h. Sodium triacetoxyborohydride (7.3 g, 34 mmol) was added at once and stirring continued overnight. Saturated aqueous NaHCO3 was added, and the resulting mixture was extracted with DCM (×2). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford a clear oil. Purification by chromatography (SiO2; hex—60% EtOAc/hex) provided the desired product as a clear oil (7.2 g, 85%). MS (ESI): mass calcd. for C16H22ClF3N2O2, 366.1; m/z found, 367.2 [M+H]+. 1H NMR (400 MHz, CDCl3) δ 7.72-7.56 (m, 2H), 7.35 (t, J=7.7 Hz, 1H), 4.94 (s, 1H), 3.99 (d, J=14.1 Hz, 1H), 3.90 (d, J=14.1 Hz, 1H), 3.29-3.14 (m, 1H), 3.11-2.99 (m, 1H), 2.84 (dd, J=11.1, 6.2 Hz, 1H), 1.44 (s, 9H), 1.11 (d, J=6.4 Hz, 3H).
Step C. (6S)-methyl 2-((1-((tert-butoxycarbonyl)amino)propan-2-yl)(2-chloro-3-(trifluoromethyl)benzyl)amino)-2-oxoacetate
-
To an ice cold solution of (6S)-tert-butyl(2-((2-chloro-3-(trifluoromethyl)benzyl)amino)propyl) carbamate (7.2 g, 20 mmol) and triethylamine (2.8 mL, 21 mmol) in DCM (121 mL) was added methyl chlorooxoacetate (1.9 mL, 21 mmol) dropwise. The resulting mixture was warmed to rt and stirred overnight. After diluting with brine, the layers were separated, and the aqueous layer washed with DCM. The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford the desired product as a white solid (8.5 g, 97%). 1H NMR (400 MHz, CDCl3) δ 7.72-7.56 (m, 1H), 7.49-7.32 (m, 2H), 4.83 (d, J=17.1 Hz, 1H), 4.79-4.62 (m, 1H), 4.51 (d, J=17.1 Hz, 1H), 4.11-3.97 (m, 1H), 3.93 (s, 3H), 3.24-3.13 (m, 2H), 1.44 (s, 9H), 1.16-1.12 (m, 3H).
Step D. (6S)-methyl 2-((1-aminopropan-2-yl)(2-chloro-3-(trifluoromethyl)benzyl)amino)-2-oxoacetate hydrochloride
-
To a solution of 4M HCl in dioxane (75 mL) was added (6S)-methyl 2-((1-((tert-butoxycarbonyl)amino)propan-2-yl)(2-chloro-3-(trifluoromethyl)benzyl)amino)-2-oxoacetate (7.5 g, 16.7 mmol). After 30 minutes, the reaction mixture was concentrated and the product was used in the next step without further purification (6.5 g, 100%). MS (ESI): mass calcd. for C14H16ClF3N2O3, 352.1; m/z found, 353.1 [M+H]+.
Step E. (6S)-1-(2-chloro-3-(trifluoromethyl)benzyl)-6-methylpiperazine-2,3-dione
-
To a solution of (6S)-methyl 2-((1-aminopropan-2-yl)(2-chloro-3-(trifluoromethyl)benzyl)amino)-2-oxoacetate hydrochloride (7.3 g, 18.9 mmol) in DCM (90 mL) was added triethylamine (7.9 mL, 57 mmol) at once. After 2 h, 1N HCl was added and the layers were separated. The aqueous layer was extracted with DCM (×2). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford the desired product as a white solid (5.9 g, 98%). MS (ESI): mass calcd. for C13H11ClF3N2O2, 320.1; m/z found, 321.1 [M+H]+. 1H NMR (600 MHz, CDCl3) δ 8.24 (d, J=3.6 Hz, 1H), 7.68 (dd, J=7.8, 1.1 Hz, 1H), 7.59 (d, J=7.7 Hz, 1H), 7.39 (t, J=7.8 Hz, 1H), 5.22 (d, J=15.7 Hz, 1H), 4.52 (d, J=15.7 Hz, 1H), 3.82-3.73 (m, 1H), 3.69-3.61 (m, 1H), 3.31 (ddd, J=13.2, 5.2, 2.3 Hz, 1H), 1.46-1.38 (m, 3H).
-
Example 14
- (±)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one………..
- ……………………(±) FORM
-

-
Example 14 was made in a manner analogous to Example 2 substituting Intermediate 3 for Intermediate 1 and 1-(bromomethyl)-2-chloro-3-(trifluoromethyl)benzene for 1-(bromomethyl)-2,3-dichlorobenzene to provide the desired compound as a white solid (102 mg, 63%). MS (ESI): mass calcd. for C18H14ClF3N6O, 422.1; m/z found, 423.1 [M+H]+. 1H NMR (500 MHz, DMSO-d6) 89.48 (d, J=1.2 Hz, 1H), 8.84-8.82 (m, 2H), 7.85-7.82 (m, 2H), 7.56 (t, J=7.8 Hz, 1H), 5.20 (d, J=16.5 Hz, 1H), 4.98 (dd, J=13.8, 2.2 Hz, 1H), 4.80 (dd, J=13.8, 4.6 Hz, 1H), 4.56 (d, J=16.6 Hz, 1H), 4.23-4.10 (m, 1H), 1.23 (d, J=6.7 Hz, 3H).
- Example 15
- (6R)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
- ……………………UNDESIRED R CONFIGURATION
-

-
Chiral SFC separation of (±)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one on a CHIRALCEL OD-H column (5 μM, 250×20 mm) using 70% CO2/30% MeOH provided 39 mg of the title compound as the first eluting enantiomer. [α]=+40° (c 2.2, CHCl3).
-
MS (ESI): mass calcd. for C18H14ClF3N6O, 422.1; m/z found, 423.1 [M+H]+. 1H NMR (500 MHz, CDCl3) δ 9.66 (d, J=1.5 Hz, 1H), 8.68 (d, J=2.5 Hz, 1H), 8.59 (dd, J=2.5, 1.5 Hz, 1H), 7.76-7.72 (m, 1H), 7.69 (dd, J=7.9, 1.6 Hz, 1H), 7.41 (t, J=7.8 Hz, 1H), 5.44 (d, J=15.5 Hz, 1H), 5.17 (dd, J=13.9, 2.1 Hz, 1H), 4.62-4.54 (m, 2H), 4.08-4.02 (m, 1H), 1.36 (d, J=6.8 Hz, 3H).
- Example 16
- (6S)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one………………
DESIRED
-

-
Chiral SFC separation of (±)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one on a CHIRALCEL OD-H column (5 μM, 250×20 mm) using 70% CO2/30% MeOH provided 40 mg of the title compound as the second eluting enantiomer.
-
[α]=−44° (c 2.2, CHCl3).
-
MS (ESI): mass calcd. for C18H14ClF3N6O, 422.1; m/z found, 423.1 [M+H]+.
-
1H NMR (500 MHz, CDCl3) δ 9.66 (d, J=1.5 Hz, 1H), 8.68 (d, J=2.5 Hz, 1H), 8.59 (dd, J=2.5, 1.5 Hz, 1H), 7.76-7.72 (m, 1H), 7.69 (dd, J=7.9, 1.6 Hz, 1H), 7.41 (t, J=7.8 Hz, 1H), 5.44 (d, J=15.5 Hz, 1H), 5.17 (dd, J=13.9, 2.1 Hz, 1H), 4.62-4.54 (m, 2H), 4.08-4.02 (m, 1H), 1.36 (d, J=6.8 Hz, 3H).
| Patent | Submitted | Granted |
|---|---|---|
| P2X7 MODULATORS [US2014275096] | 2014-03-14 | 2014-09-18 |

//////////////P2X7, 6,7-Dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one, Autoradiography, Depression, CNS, Preclinical characterization, substituted 6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one, P2X7 receptor antagonists, Janssen Pharmaceutical Research & Development L.L.C, 1627748-32-6
FC(F)(F)c4cccc(CN1C(=O)c2nnc(n2C[C@@H]1C)c3cnccn3)c4Cl
CC1CN2C(=NN=C2C(=O)N1CC3=C(C(=CC=C3)C(F)(F)F)Cl)C4=NC=CN=C4