
Nemonoxacin 奈诺沙星
378746-64-6 CAS
TG-873870
- C20-H25-N3-O4
- 371.4345
WARNER CHILCOTT ORIGINATOR
CLINICAL TRIALS http://clinicaltrials.gov/search/intervention=Nemonoxacin
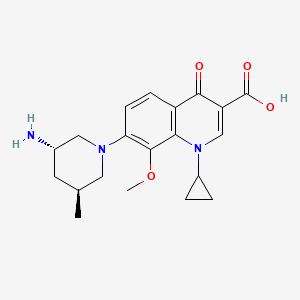
(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
7-[3(S)-Amino-5(S)-methylpiperidin-1-yl]-1-cyclopropyl-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Taigexyn has been approved in Taiwan IN 2014
Taigexyn has been approved in Taiwan IN 2014
"TAIPEI, MARCH 13, 2014 /PRNEWSWIRE/ -- TAIGEN BIOTECHNOLOGY ..."
13.03.14 |
13.03.14 |
TaiGen Biotechnology Receives Marketing Approval from the Taiwan Food and Drug Administration for Taigexyn in Taiwan
TAIPEI, March 13, 2014 /PRNewswire/ -- TaiGen Biotechnology Company, Limited ("TaiGen") today announced that the Taiwan Food and Drug Administration (TFDA) has approved the new drug application (NDA) of Taigexyn® (nemonoxacin) oral formulation (500 mg) for the treatment of community-acquired bacterial pneumonia (CAP). With this NDA approval, Taiwan is the first region to grant marketing approval to Taigexyn®. An NDA for Taigexyn® was also submitted to China FDA (CFDA) in April 2013 and is currently under review.
Nemonoxacin is a novel non-fluorinated quinolone antibiotic undergoing clinical trials.
Taigexyn Granted QIDP and Fast Track Designations
TaiGen Biotechnology announced that the FDA has granted nemonoxacin (Taigexyn) Qualified Infectious Disease Product (QIDP) and Fast Track designations for community-acquired bacterial pneumonia (CAP) and acute bacterial skin and skin structure infections (ABSSSI).
Nemonoxacin is a novel non-fluorinated quinolone broad spectrum antibiotic available in both oral and intravenous formulations. Nemonoxacin demonstrates activity against gram-positive and gram-negative bacteria and atypical pathogens. Nemonoxacin also possesses activities against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant pathogens.
Nemonoxacin is a novel non-flourinated quinolone antibiotic registered in Taiwan for the oral treatment of community-acquired pneumonia. Clinical trials are in development at TaiGen Biotechnology for the treatment of diabetic foot infections and for the treatment of moderate to severe community-acquired pneumonia with an intravenous formulation. The drug is thought to accomplish its antibacterial action through topoisomerase inhibition.
Originally developed at Procter & Gamble, nemonoxacin was the subject of a strategic alliance formed in January 2005 between P&G and TaiGen to further the development and commercialization of nemonoxacin. In 2012, the product was licensed by TaiGen Biotechnology to Zhejiang Medicine in China for manufacturing, sales and marketing. In 2014, TaiGen out-licensed the exclusive rights of the product in Russian Federation, Commonwealth Independent States and Turkey to R-Pharm.
TaiGen has completed two Phase 2 clinical studies, one in CAP and the other in diabetic foot infections with demonstrated efficacy and safety. In the clinical trials conducted to date, nemonoxacin has shown activity against drug-resistant bacteria such as MRSA, quinolone-resistant MRSA, as well as quinolone-resistant Streptococcus pneumoniae.

Malate salt
Chemical structure of nemonoxacin as a malate salt (C20H25N3O4·C4H6O5·H2O). Nemonoxacin is the free base, and its molecular mass is 371.44 g/mol. The molecular mass of the salt, nemonoxacin malate, is 514.53 g/mol.


..........................
isomeric compounds are:

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD1.......DESIRED

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1'....NOT DESIRED
Example 1
Malate salts of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and (3S,5R)-7- [3-ammo-5-methyl-piperidinyl]- 1 -cyclopropyl- 1 ,4-dihydro-8-methoxy-4-oxo-3- quinolinecarboxylic acid (Compound 1') were synthesized as follows:
(A) Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9) and (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9'): Compound 9' was synthesized as shown in Scheme 1 below:
Scheme 1

3 4 Boc

A 50-L reactor was charged with Compound 2 (5.50 kg, 42.60 mol), methanol (27 L) and cooled to 10-150C. Thionyl chloride (10.11 kg, 2.0 equiv.) was added via an addition funnel over a period of 65 min, with external cooling to keep temperature below 30°. The resulting solution was stirred at 250C for 1.0 hour, after which methanol was removed under reduced pressure. The oily residue was azeotroped with ethyl acetate (3 x 2.5 L) to remove residual methanol, dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by slow addition of triethylamine (3.6 kg) below 3O0C. The resulting suspension was filtered to remove triethylamine hydrochloride.
The filtrate was charged to a 50 L reactor, along with DMAP (0.53 kg). Di- fert-butyl dicarbonate (8.43 kg) was added via hot water heated addition funnel, over a period of 30 min at a temperature of 20-300C. The reaction was complete after 1 hour as determined by TLC analysis. The organic phase was washed with ice cold IN HCl (2 x 7.5 L), saturated sodium bicarbonate solution (1 x 7.5 L), dried over magnesium sulfate, and filtered. After ethyl acetate was removed under reduced pressure, crystalline slurry was obtained, triturated with MTBE (10.0 L), and filtered to afford Compound 3 as a white solid (5.45 kg, 52.4%).
Anal. Calcd for CHHI7NO5 : C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for CHHI8NO5, [M+H] 244.1185. Found
244.1174; 1H NMR (CDCl3, 500 MHz):δ=4.54 (dd, J= 3.1, 9.5 Hz, IH), 3.7 (s, 3H), 2.58-2.50 (m, IH), 2.41 (ddd, IH, J= 17.6, 9.5, 3.7), 2.30-2.23 (m, IH), 1.98-1.93 (m, IH), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5. Mp 70.20C.
A 50-L reactor was charged with Compound 3 (7.25 kg, 28.8 mol), DME (6.31 kg), and Bredereck's Reagent (7.7 kg, 44.2 mole). The solution was agitated and heated to 750C + 50C for three hours. The reaction was cooled to O0C over an hour, during which time a precipitate formed. The mixture was kept at O0C for an hour, filtered, and dried in a vacuum oven for at least 30 hours at 3O0C + 50C to give compound 4 as a white crystalline solid (6.93 kg, 77.9%).
Anal. Calcd for Ci4H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for Ci4H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR (CDCl3, 499.8 MHz) δ = 7.11 (s, IH), 4.54 (dd, IH, J= 10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, IH), 3.00 (s, 6H), 2.97-2.85 (m,lH), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ = 172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. MP 127.90C. A 10-gallon Pfaudler reactor was charged with ESCAT 142 (Engelhard Corp.
N.J, US) 5% palladium powder on carbon (50% wet, 0.58 kg wet wt), Compound 4 (1.89 kg, 6.33 mol), and isopropanol (22.4 Kg). After agitated under a 45-psi hydrogen atmosphere at 450C for 18 hrs, the reaction mixture was cooled to room temperature and filtered though a bed of Celite (0.51 kg). The filtrate was evaporated under reduced pressure to give a thick oil, which was solidified on standing to afford Compound 5 (1.69 kg, 100%) as a 93:7 diastereomeric mixture.
A sample of product mixture was purified by preparative HPLC to give material for analytical data. Anal. Calcd for Ci2Hi9NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for Ci2Hi9NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ = 4.44 (m, IH), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, IH), 1.43 (s, 9H), 1.20 (d, j = 6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ = 175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.90C.
A 50-L reactor was charged with Compound 5 (3.02 kg, 11.7 mol), absolute ethanol (8.22 kg), and MTBE (14.81 kg). Sodium borohydride (1.36 kg, 35.9 mol) was added in small portions at 00C + 50C. A small amount of effervescence was observed. The reaction mixture was warmed to 1O0C + 50C and calcium chloride dihydrate (2.65 kg) was added in portions at 1O0C + 50C over an hour. The reaction was allowed to warm to 2O0C + 50C over one hour and agitated for an additional 12 hours at 200C + 50C. After the reaction was cooled to -50C + 50C, ice-cold 2N HCl (26.9 kg) was added slowly at of O0C + 50C. Agitation was stopped. The lower aqueous phase was removed. The reactor was charged with aqueous saturated sodium bicarbonate (15.6 kg) over five minutes under agitation. Agitation was stopped again and the lower aqueous phase was removed. The reactor was charged with magnesium sulfate (2.5 kg) and agitated for at leastlO minutes. The mixture was filtered though a nutsche filter, and concentrated under reduced pressure to afford Compound 6 (1.80 kg, 66%). Anal. Calcd for CnH23NO4: C, 56.6 H, 9.94; N, 6.00. Found C, 56.0; H, 9.68;
N, 5.96; HRMS (ESI+) Expected for CnH24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz) δ = 6.34 (d, J= 8.9 Hz, IH, NH), 4.51 (t, J= 5.8, 5.3 Hz, IH, NHCHCH2OH), 4.34 (t, J= 5.3, 5.3 Hz, IH, OBCHCH2OH), 3.46-3.45, (m, IH, NHCH), 3.28 (dd, J= 10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J= 10.2, 5.8 Hz , IH, CH3CHCHHOH), 3.16 (dd, J = 10.2, 6.2 Hz, IH, NHCHCHHOH), 3.12 (dd, J= 10.6, 7.1 Hz , IH, CH3CHCHHOH), 1.53-1.50 (m, IH, CH3CHCHHOH), 1.35 (s, 9H, 0(CHB)3, 1.30 (ddd, J = 13.9, 10.2, 3.7 Hz, IH, NHCHCHHCH), 1.14 (ddd, J= 13.6, 10.2, 3.4 Hz, IH, NHCHCHHCH), 0.80 (d, J= 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.10C. A 50 L reactor was charged with a solution of Compound 6 (5.1 kg) in isopropyl acetate (19.7 kg). The reaction was cooled to 150C + 5°C and triethylamine (7.8 kg) was added at that temperature. The reactor was further cooled to O0C + 50C and methanesulfonyl chloride (MsCl) (6.6 kg) was added. The reaction was stirred for a few hours and monitored for completion by HPLC or TLC. The reaction was quenched by saturated aqueous bicarbonate solution. The organic phase was isolated and washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase was dried, filtered, and concentrated in vacuo below 550C + 50C to afford compound 7 as a solid/liquid slurry, which was used in the subsequent reaction without further purification.
After charged with 9.1 kg of neat benzylamine, a 50 L reactor was warmed to 550C, at which temperature, a solution of compound 7 (8.2 kg) in 1,2- dimethoxyethane (14.1 kg) was added. After the addition, the reaction was stirred at 6O0C + 50C for several hours and monitored for completion by TLC or HPLC. The reaction was cooled to ambient temperature and the solvent was removed under vacuum. The residue was diluted with 11.7 kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture was obtained upon standing. The upper organic layer was collected. The isolated middle layer was extracted twice again with 11.7 kg portions of 15% (v/v) ethyl acetate/hexanes solution. The combined organic layers were concentrated under vacuum to give an oily residue. The residue was then purified by chromatography to afford Compound 8 as an oil. A 40 L pressure vessel was charged with 0.6 kg 50% wet, solid palladium on carbon (ElOl, 10 wt. %) under flow of nitrogen. A solution of Compound 8 (3.2 kg) in 13.7 kg of absolute ethanol was then added to the reactor under nitrogen. The reactor was purged with nitrogen and then pressurized with hydrogen at 45 psi. The reaction was then heated to 45°C. It was monitored by TLC or LC. Upon completion, the reaction was cooled to ambient temperature, vented, and purged with nitrogen. The mixture was filtered through a bed of Celite and the solid was washed with 2.8 kg of absolute ethanol. The filtrate was concentrated under vacuum to afford Compound 9 as a waxy solid.
TLC R/(Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain) = 0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, IH), 3.80-3.68 (m, IH), 2.92 (d, J=I 1.4 Hz,
IH), 2.77 (AB quart, JAB=12.0 Hz, v=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, IH), 1.82-1.68 (m, 2H), 1.54 (br s, IH), 1.43 (s, 9H), 1.25-1.15 (m, IH), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H). Similarly, (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester
(Compound 9') was synthesized as shown in Scheme 2.
Scheme 2

HN Boc HN Boc
NaBH4,EtOH w - " MsCI1TEA . „ _. - - _. „ Benzyl Amine
THF EA1CoId

(B) Synthesis of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3- carboxylic acid (Compound 10): Compound 10 was prepared according to the method described in U.S. Patent
6,329,391.
(C) Synthesis of borone ester chelate of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo- l,4-dihydro-quinoline-3-carboxylic acid (Compound 11):
Scheme 3

Toluene, tert-Butylmethyl ether 20-500C, filter
A reactor was charged with boron oxide (2.0 kg, 29 mol), glacial acetic acid (8.1 L, 142 mol), and acetic anhydride (16.2 L, 171 mol). The resulting mixture was refluxed at least 2 hours, and then cooled to 400C, at which temperature, 7- fluoroquinolone acid compound 10 (14.2 kg, 51 mol) was added. The mixture was refluxed for at least 6 hours, and then cooled to about 900C. Toluene (45 L) was added to the reaction. At 5O0C, terϊ-butylmethyl ether (19 L) was added to introduce precipitation. The mixture was then cooled to 200C and filtered to isolate the precipitation. The isolated solid was then washed with teτt-butylmethyl ether (26 L) prior to drying in a vacuum oven at 4O0C (50 torr) to afford Compound 11 in a yield of 86.4%. Raman (cm 1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, IH), 8.38-8.33 (m, IH), 7.54 (t, J=9.8 Hz, IH), 4.38-4.35 (m, IH), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 6θA, 200 μm), Mobile Phase: 1 :1 (v/v) CH3CN : 0.5N NaCl (aq), UV (254/366 nm) visualization; R^O.4-0.5. (D) Synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidmyl]-l- cyclopropyl-l,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and malate salt of (3S,5R)-7-[3-amino-5-methyl-piperidmyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1')
Compound 1 was synthesized from compound 9 as shown in Scheme 4 below:
Scheme 4

5O0C 3 d
a 6 0 N HCI (aq) CH2CI2 35°40°C 12 h t> Extract pH ad]ust to ~7-8 50"-65"C filter



A reactor was charged with Compound 11 (4.4 kg, 10.9 mol), Compound 9 (2.1 kg, 9.8 mol), triethylamine (TEA) (2.1 L, 14.8 mol), and acetonitrile (33.5 L, 15.7 L/kg). The resulting mixture was stirred at approximately 500C till completion of the reaction, as monitored by HPLC or reverse phase TLC. It was cooled to approximately 35°C and the reaction volume was reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. After 28.2 kg of 3.0 N NaOH (aq) solution was added, the reaction mixture was warmed to approximately 4O0C, distilled under vacuum until no further distillates were observed, and hydro lyzed at room temperature. Upon completion of hydrolysis, which was monitored by HPLC or reverse phase TLC, 4-5 kg of glacial acetic acid was added to neutralize the reaction mixture.
The resulting solution was extracted 3 times with 12.7 kg (9.6 L) of dichloromethane. The organic layers were combined and transferred to another reactor. The reaction volume was reduced to approximately a half by evaporation at 400C. After 20.2 Kg 6.0N HCl (aq) solution was added, the reaction mixture was stirred for at least 12 hours at 35°C. After the reaction was completed as monitored by HPLC or reverse phase TLC, agitation was discontinued to allow phase separation. The organic phase was removed and the aqueous layer was extracted with 12.7 kg (9.6 L) of dichloromethane. The aqueous layer was diluted with 18.3 kg distilled water and warmed to approximately 500C. Dichloromethane was further removed by distillation under vacuum (100-400 torr).
The pH of the aqueous solution was then adjusted to 7.8-8.1 by adding about 9.42 kg of 3.0 N NaOH (aq) below 65°C. The reaction mixture was stirred at 500C for at least an hour and then cooled to room temperature. The precipitate was isolated by suction filtration, washed twice with 5.2 kg of distilled water, and dried with suction for at least 12 hours and then in a convection oven at 55°C for additional 12 hours. Compound 12 (3.2 kg, 79%) was obtained as a solid.
A reactor was charged with 3.2 kg of Compound 12 and 25.6 kg of 95% ethanol. To the reactor was added 1.1 kg of solid D,L-malic acid. The mixture was refluxed temperature (~80°C). Distilled water (-5.7 L) was added to dissolve the precipice and 0.2 kg of activated charcoal was added. The reaction mixture was passed through a filter. The clear filtrate was cooled to 45°C and allowed to sit for at least 2 hours to allow crystallization. After the reaction mixture was further cooled to 5°C, the precipitate was isolated by suction filtration, washed with 6.6 kg of 95% ethanol, and dried with suction for at least 4 hours. The solid was further dried in a convection oven at 450C for at least 12 hours to afford 3.1 kg of Compound 1 (yield: 70%). NEMONOXACIN
NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, IH), 7.37 (d, J=9.0 Hz, IH), 7.05 (d, J=9.0 Hz, IH), 4.23-4.18 (m, IH), 4.10-3.89 (m, IH), 3.66 (br s, IH), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, IH), 3.34 (d, J=9.3 Hz, IH), 3.16 (d, J=12.9 Hz, IH), 2.65 (dd, J=16.1, 4.1 Hz, IH), 2.64-2.53 (m, IH), 2.46 (dd, J=16.1, 8.0 Hz, IH), 2.06 (br s, IH), 1.87 (d, J=14.4 Hz, IH), 1.58-1.45 (m, IH), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H), 0.85-0.78 (m, 2H).
Similarly, Compound 1' was synthesized from Compound 9' as shown in Scheme 5 below:
Scheme 5

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1'....NOT DESIRED
.....................
US2007/232650 A1,
malate salts of

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (hereinafter Compound I, see also intermediate (23) in Section D, of Detailed Description of the Invention).
EXAMPLES Example 1 Synthesis of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid and malate salt thereof A. Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8)

(2S)-1-(1,1-Dimethylethyl)-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester, (2). A 50-L reactor is charged with compound (1) (5.50 Kg, 42.60 mol), methanol (27 L) and cooled to 10-15° C. Thionyl chloride (10.11 Kg, 2.0 equiv.) is added via addition funnel over a period of 65 min, with external cooling to maintain temperature at <30°. The resulting solution is stirred at 25° C.+5° C. for 1.0 hour, after which the methanol is distilled off under reduced pressure. The resulting thick oil is azeotroped with ethyl acetate (3×2.5 L) to remove residual methanol. The residue is dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by the addition of triethylamine (3.6 Kg) from an addition funnel over 30 minutes. The temperature of the neutralization is maintained below 30° C. via external cooling. The resulting suspension of triethylamine hydrochloride is removed by filtration, and the clarified mother liquor solution is charged to a 50 L reactor, along with DMAP (0.53 Kg). Di-tert-butyl dicarbonate (8.43 Kg) is added via hot water heated addition funnel, over a period of 30 min with external cooling to maintain temperature at about 20-30° C. The reaction is complete after 1 hour as determined by TLC analysis. The organic phase is washed with ice cold 1N HCl (2×7.5 L), saturated sodium bicarbonate solution (1×7.5 L), and dried over magnesium sulfate. The mixture is filtered through a nutsche filter and ethyl acetate is removed under reduced pressure to yield a crystalline slurry that is triturated with MTBE (10.0 L) and filtered to afford intermediate (2) as a white solid (5.45 Kg, 52.4%). Anal. Calcd for C11H17NO5: C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for C11H18NO5, [M+H] 244.1185. Found 244.1174; 1H NMR (CDCl3, 500 MHz): δ=4.54 (dd, J=3.1, 9.5 Hz, 1H), 3.7 (s, 3H), 2.58-2.50 (m, 1H), 2.41 (ddd, 1H, J=17.6, 9.5, 3.7), 2.30-2.23 (m, 1H), 1.98-1.93 (m, 1H), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5; Mp 70.2° C.
(2S,4E)-1-(1,1-Dimethylethyl)-4-[(dimethylamino)methylene]-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (3). A 50-L reactor is charged with intermediate (2) (7.25 Kg, 28.8 mol), DME (6.31 Kg), and Bredereck's Reagent (7.7 Kg, 44.2 mole). The solution is agitated and heated to 75° C.±5° C. for at least three hours. The progress of the reaction is monitored by HPLC. The reaction is cooled to 0° C.±5° C. over on hour during which time a precipitate forms. The mixture is held at 0° C.±5° C. for one hour and filtered though a nutsche filter and the product dried in a vacuum oven for at least 30 hours at 30° C.±5° C. to give intermediate (3) as a white crystalline solid (6.93 Kg, 77.9%). Anal. Calcd for C14H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for C14H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR(CDCl3, 499.8 MHz)δ=7.11 (s, 1H), 4.54 (dd, 1H, J=10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, 1H), 3.00 (s, 6H), 2.97-2.85 (m, 1H), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ=172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. Mp 127.9° C.
(2S,4S)-1-(1,1-Dimethylethyl)-4-methyl-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (4). A 10-gallon Pfaudler reactor is inerted with nitrogen and charged with ESCAT 142 5% palladium powder on carbon (50% wet, 0.58 Kg wet wt.), intermediate (3) (1.89 Kg, 6.33 mol) and isopropanol (22.4 Kg). The reaction mixture is agitated under a 45-psi hydrogen atmosphere at 45° C. for 18 hrs. The reaction mixture is then cooled to room temperature and filtered though a bed of Celite (0.51 Kg) in a nutsche filter to remove catalyst. The mother liquor is evaporated under reduced pressure to give a thick oil that crystallizes on standing to afford 4 (1.69 Kg, 100%) as a 93:7 diastereomeric mixture. A sample of product mixture is purified by preparative HPLC to give material for analytical data. Anal. Calcd for C12H19NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for C12H19NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ=4.44 (m, 1H), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, 1H), 1.43 (s, 9H), 1.20 (d, j=6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ=175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.9° C.
(1S,3S)-(4-Hydroxyl-1-hydroxymethyl-3-methyl-butyl)-carbamic acid tert-butyl ester (5). A 50-L reactor is charged with intermediate (4) (3.02 Kg, 11.7 mol), absolute ethanol (8.22 Kg), and MTBE (14.81 Kg). The solution is agitated and cooled to 0° C.±5° C. and sodium borohydride (1.36 Kg, 35.9 mol) is added in small portions so as to maintain reaction temperature at 0° C.±5° C. A small amount of effervescence is observed. The reaction mixture is warmed to 10° C.±5° C. and calcium chloride dihydrate (2.65 Kg) is added portion wise at a slow rate over an hour so as to maintain a reaction temperature of 10° C.±5° C. The reaction is allowed to warm to 20° C.±5° C. over one hour and agitated for an additional 12 hours at 20° C.±5° C. The reaction is cooled to −5° C.±5° C., ice-cold 2N HCl (26.9 Kg) is added at a rate to maintain a reaction temperature of 0° C.±5° C. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=1) is removed. The reactor is charged with aqueous saturated sodium bicarbonate (15.6 Kg) over five minutes. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=8) is removed. The reactor is charged with magnesium sulfate (2.5 Kg) and agitated for at least 10 minutes. The mixture is filtered though a nutsche filter, and condensed under reduced pressure to afford intermediate (5) (1.80 Kg, 66%). Anal. Calcd for C11H23NO4: C, 56.6; H, 9.94; N, 6.00. Found C, 56.0; H, 9.68; N, 5.96; HRMS (ESI+) Expected for C11H24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz)δ=6.34(d, J=8.9 Hz, 1H, NH), 4.51 (t, J=5.8, 5.3 Hz, 1H, NHCHCH2OH), 4.34 (t, J=5.3, 5.3 Hz, 1H, CH3CHCH2OH), 3.46-3.45, (m, 1H, NHCH), 3.28 (dd, J=10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J=10.2, 5.8 Hz, 1H, CH3CHCHHOH), 3.16 (dd, J=10.2, 6.2 Hz, 1H, NHCHCHHOH), 3.12 (dd, J=10.6, 7.1 Hz, 1H, CH3CHCHHOH), 1.53-1.50 (m, 1H, CH3CHCHHOH), 1.35 (s, 9H, O(CH 3)3, 1.30 (ddd, J=13.9, 10.2, 3.7 Hz, 1H, NHCHCHHCH), 1.14 (ddd, J=13.6, 10.2, 3.4 Hz, 1H, NHCHCHHCH), 0.80 (d, J=6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.1° C.
(2S,4S)-Methanesulfonic acid 2-tert-butoxycarbonylamino-5-methanesulfonyloxy-4-methyl-pentyl ester (6). A 50 L reactor is charged with a solution of intermediate (5) (5.1 Kg) in isopropyl acetate (i-PrOAc) 11.8 Kg followed by a rinse with an additional 7.9 Kg i-PrOAc. The reaction is cooled to 15° C.±5° C. and triethylamine (TEA) (7.8 Kg) is added while maintaining the set temperature. The reactor is further cooled to 0° C.±5° C. and methanesulfonyl chloride (MsCl) (6.6 Kg) is added to the reaction solution while maintaining the set temperature. The reaction is stirred for a few hours and monitored for completion by HPLC or TLC. The reaction is quenched by the addition of a saturated aqueous bicarbonate solution and the resulting isolated organic phase is washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase is dried, filtered, and concentrated in vacuo below 55° C.±5° C. until a solid/liquid slurry containing intermediate (6) is obtained. The slurry is used crude in subsequent reaction without further characterization.
(3S,5S)-(1-Benzyl-5-methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (7). A 50 L reactor is charged with 9.1 Kg of neat benzylamine. The reactor is brought to 55° C. and a solution of intermediate (6) (8.2 Kg) in 1,2-dimethoxyethane (DME) (14.1 Kg) is added to the reactor while maintaining a temperature of 60° C.±5° C. After complete addition of this solution, the reaction is stirred at 60° C.±5° C. for several hours and monitored for completion by TLC or HPLC. The reaction is cooled to ambient temperature and volatiles (DME) are removed by rotary evaporation under vacuum. The residue is diluted with 11.7 Kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 Kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture is obtained upon settling. The bottom aqueous phase is removed and the middle phase is set aside. The upper organic phase is collected and held for combination with extracts from additional extractions. The isolated middle phase is extracted twice again with 11.7 Kg portions of 15% (v/v) ethyl acetate/hexanes solution, each time combining the extracts with original organic phase. The combined organic extracts are transferred into a rotary evaporator and solvent is removed under vacuum until an oily residue remains. The residue is then purified via large-scale preparative chromatography to afford purified intermediate (7) as an oil.
(3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8). A 40 L pressure vessel is charged with 0.6 Kg 50% wet, solid palladium on carbon (E101, 10 wt. %) under flow of nitrogen. A solution of 3.2 Kg intermediate (7) in 13.7 Kg of absolute ethanol is then charged to the reactor under nitrogen. The reactor is purged with nitrogen and is then pressurized with hydrogen at 45 psi. The reaction is then heated to 45° C. while maintaining a hydrogen pressure of 45 psi. The reaction is monitored by TLC or LC until complete. The reaction is cooled to ambient temperature, vented, and purged with nitrogen. The reactor contents are filtered through a bed of Celite and the solids are washed with 2.8 Kg of absolute ethanol. The filtrate is concentrated by rotary evaporation under vacuum until a waxy solid is obtained to afford intermediate (8): TLC Rf (Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain)=0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, 1H), 3.80-3.68 (m, 1H), 2.92 (d, J=11.4 Hz, 1H), 2.77 (AB quart, JAB=12.0 Hz, Δν=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, 1H), 1.82-1.68 (m, 2H), 1.54 (br s, 1H), 1.43 (s, 9H), 1.25-1.15 (m, 1H), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H).
B. Synthesis of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (19)


Intermediate (12): A reactor is charged with a solution of intermediate (11) (1.2 Kg, 7.7 mol, 1.0 eq) in anhydrous toluene (12 L) followed by ethylene glycol (1.8 L, 15.7 mol, 4.2 eq) and solid p-toluenesulfonic acid (120 g, 10 wt. %). The reaction mixture is stirred at ambient temperature for at least 30 minutes and then heated to reflux, collecting the water/toluene azeotrope in a Dean Stark type trap apparatus until the reaction is complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to ambient temperature and poured into an aqueous solution of sodium bicarbonate (6 L). The organic toluene phase was removed and washed with saturated sodium bicarbonate solution (6 L), distilled water (2×6 L), and saturated aqueous brine (6 L). The organic phase was removed and dried over MgSO4, filtered, and evaporated under reduced pressure to afford intermediate (12) as an oil (1.3 Kg, 86%). The material is used without further purification in subsequent reaction steps.
Intermediate (13): A reactor is charged with a solution of intermediate (12) (1.2 Kg, 6.0 mol, 1.0 eq) in anhydrous tetrahydrofuran (12 L) and n-butyllithium (2.5M in hexanes, 2.6 L, 6.6 mol, 1.1 eq) is added at −40° C., while maintaining this temperature throughout the addition. The reaction is stirred for at least one hour at −40° C. and trimethylborate (0.9 L, 7.8 mol, 1.3 eq) is added to the mixture while maintaining the temperature at or below −40° C. The reaction mixture is stirred for at least one hour at −40° C. until complete as determined by TLC analysis (30% EtOAc/Hexanes v/v). The reaction is warmed slightly to −30° C. and acetic acid (3 L) is added slowly. Upon complete addition, water is added (0.5 L) to the reaction and the mixture is allowed to quickly warm to ambient temperature while stirring overnight. Organic solvent is removed from the reaction by distillation under reduced pressure at 45° C. To the reaction residue is added 3-4 volumes of water (6 L) and 30% hydrogen peroxide (0.7 L, 1.0 eq) slowly at ambient temperature with cooling provided to control the exotherm. The reaction is stirred for at least an hour at ambient temperature until complete as determined by TLC (15% EtOAc/Hexanes v/v). The reaction mixture is cooled to 0-5° C. and excess peroxide is quenched with the addition of 10% aqueous sodium bisulfite solution (2 L). The mixture is tested to ensure a negative peroxide result and the reaction is acidified by the addition of 6N HCl (aq) (1.2 L). The reaction is stirred until the hydrolysis reaction is complete as determined by TLC or NMR analysis. The resulting solids are collected by suction filtration to afford intermediate (13) as a yellow solid (1.0 Kg, 79%).
Intermediate (14): A reactor is charged with intermediate (13) (0.53 Kg, 3.0 mol, 1.0 eq) and dissolved in dry toluene (2.7 Kg, 3.1 L). To this solution is added dimethylsulfate (0.49 Kg, 3.9 mol, 1.30 eq) followed by solid potassium carbonate (0.58 Kg, 4.2 mol, 1.4 eq). The reaction mixture is heated to reflux and held for at least 1 hour until complete as determined by HPLC. During this time, vigorous gas evolution is observed. The reaction is then cooled to ambient temperature and diluted with distilled water (3.2 L) along with 30% NaOH (aq) (0.13 Kg, 0.33 eq). The aqueous phase is separated and the remaining toluene phase is extracted twice more with distilled water (3.2 L) combined with 30% NaOH (aq) (0.13 Kg, 0.33 eq), removing the aqueous phase each time. The organic upper phase is concentrated by distillation in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature, checked for quality and yield by HPLC, and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (14) assumed, 0.56 Kg).
Intermediate (15a,b): A reactor is charged with 1.8 Kg (2.1 L) anhydrous toluene along with sodium hydride (0.26 Kg, 6.6 mol, 2.20 eq) as a 60 wt. % dispersion in mineral oil. To this mixture is added (0.85 Kg, 7.2 mol, 2.4 eq) diethylcarbonate as the reaction mixture is heated to 90° C. over 1 hour. A solution of intermediate (14) (˜1.0 eq) in toluene from the previous step is added to the reaction while maintaining a temperature of 90° C.±5° C. Gas evolution can be observed during this addition. After complete addition, the reaction is stirred for at least 30 minutes or until complete as determined by HPLC analysis. Upon completion, the mixture is cooled to ambient temperature and diluted with 10 wt. % aqueous sulfuric acid (3.8 Kg, 3.9 mol, 1.3 eq) with agitation. The phases are allowed to separate and the lower aqueous phase is removed. The remaining organic phase is concentrated in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (15a,b) assumed, 0.85 Kg).
Intermediate (16a,b; 17a,b): A reactor is charged with a solution of intermediate (15a,b) (0.85 Kg, ˜3.0 mol, ˜1.0 eq) in toluene from the previous step. To the reactor is then added dimethylformamide-dimethylacetal (0.54 Kg, 4.5 mol, 1.5 eq) and the resulting solution is heated to reflux temperature (˜95-105° C.). The lower boiling solvent (methanol from reaction) is allowed to distill off while the temperature is maintained at ≧90° C. Heating is continued for at least 1 hour or until complete as determined by HPLC analysis. Upon completion, the reaction containing the mixture of intermediate (16a,b), is cooled to ambient temperature and toluene (1.8 Kg, 2.1 L) along with cyclopropylamine (0.21 Kg, 3.6 mol, 1.2 eq) are added to the reaction. The reaction is stirred at ambient temperature for at least 30 minutes until complete as determined by HPLC. Upon completion, the reaction is diluted with 10 wt. % aqueous sulfuric acid (2.9 Kg, 3.0 mol, 1.0 eq) with agitation, and the phases are then allowed to separate. The aqueous phase is removed and the organic phase is concentrated under reduced pressure (<100 mbar) at approximately 40° C. by distillation. When the desired concentration is achieved, the solution is cooled to ambient temperature and the toluene solution containing the mixture of intermediate (17a,b) is carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (17a,b) assumed, ˜1.1 Kg).
Intermediate (18): A reactor is charged with a solution of the mixture of intermediate (17a,b) (˜4.7 Kg, ˜3.0 mol) at ambient temperature. To the reactor is added N,O-bis(trimethylsilyl)acetamide (0.61 Kg, 3.0 mol, 1.0 eq) and the reaction is heated to reflux temperature (˜105-115° C.) for at least 30 minutes or until complete as determined by HPLC analysis. If not complete, an additional amount of N,O-bis(trimethylsilyl)acetamide (0.18 Kg, 0.9 mol, 0.3 eq) is added to the reaction to achieve completion. Upon completion, the reaction is cooled to below 40° C. and organic solvent is removed under reduced pressure (<100 mbar) at approximately 40° C. by distillation until a precipitate is formed. The reaction is cooled to ambient temperature and the precipitated solids are isolated by suction filtration and washed with distilled water twice (1×1.8 L, 1×0.9 L). The solid is dried to afford intermediate (18) as a white solid (0.76 Kg, 82%). The material is used without further purification in the next reaction step.
Intermediate (19): A reactor is charged with solid intermediate (18) (0.76 Kg, ˜2.5 mol, ˜1.0 eq) at ambient temperature followed by ethanol (5.3 Kg, 6.8 L) and 32 wt. % aqueous hydrochloric acid (1.1 Kg, 10 mol). The reaction mixture is brought to reflux temperature (76-80° C.) during which time the mixture first becomes homogeneous and later becomes heterogeneous. The mixture is heated at reflux for at least 5 hours or until complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to 0° C.±5° C. and the precipitated solid is isolated by filtration and washed with distilled water (1.7 Kg) followed by ethanol (1.7 Kg). The isolated solid is dried to afford intermediate (19) as a white solid (0.65 Kg, ˜95%). 1H NMR (CDCl3, 300 MHz) δ (ppm): 14.58 (s, 1H), 8.9 (s, 1H), 8.25 (m, 1H), 7.35 (m, 1H), 4.35 (m, 1H), 4.08 (s, 3H), 1.3 (m, 2H), 1.1 (m, 2H) 19F NMR (CDCl3+CFCl3, 292 MHz) δ (ppm): −119. HPLC: 99.5% by area.
C. Synthesis of borone ester chelate of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20)

A reactor is charged with boron oxide (2.0 Kg, 29 mol) followed by dilution with glacial acetic acid (8.1 L, 142 mol) and acetic anhydride (16.2 L, 171 mol). The resulting mixture is heated to reflux temperature for at least 2 hours. The reaction contents are cooled to 40° C. and the solid 7-fluoroquinolone acid intermediate (19) (14.2 Kg, 51 mol) is added to the reaction mixture. The mixture is again heated to reflux temperature for at least 6 hours. Reaction progress is monitored by HPLC and NMR. The mixture is cooled to approximately 90° C. and toluene (45 L) is added to the reaction. The reaction is further cooled to 50° C. and tert-butylmethyl ether (19 L) is added to the reaction mixture to bring about precipitation of the product. The mixture is then cooled to 20° C. and the solid product 19 is isolated by filtration. The isolated solids are then washed with tert-butylmethyl ether (26 L) prior to drying in a vacuum oven at 40° C. (50 torr). The product yield obtained for intermediate (20) in this reaction is 86.4%. Raman (cm−1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, 1H), 8.38-8.33 (m, 1H), 7.54 (t, J=9.8 Hz, 1H), 4.38-4.35 (m, 1H), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization; Rf=0.4-0.5.
D. Coupling of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20) to (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8), and synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (25)

A reactor is charged with solid intermediate (20) (4.4 Kg, 10.9 mol) followed by dilution with a solution of triethylamine (TEA) (2.1 L, 14.8 mol) and piperidine side chain intermediate (8) (2.1 Kg, 9.8 mol) in acetonitrile (33.5 L, 15.7 L/Kg) at room temperature. The resulting mixture is warmed to approximately 50° C. until reaction is judged complete. Reaction progress is monitored by HPLC or reverse phase TLC. When complete, the reaction is cooled to approximately 35° C. and reaction volume is reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. The reactor is then charged with 28.2 Kg of 3.0N NaOH (aq) solution and the temperature is raised to approximately 40° C. Distillation under vacuum is continued between 1-4 hours or until no further distillates are observed. The reaction is then cooled to room temperature and the hydrolysis reaction is monitored by HPLC or reverse phase TLC. Upon completion, the reaction mixture is neutralized to a pH of between 6-8 by adding ˜4-5 Kg of glacial acetic acid. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The extraction process is repeated two additional times using 12.7 Kg (9.6 L) of dichloromethane, collecting the lower, organic phase each time. The aqueous phase is discarded and the organic extracts are combined in a single reactor. The reactor contents are heated to 40° C. and the reaction volume is reduced to approximately one half by distillation. The reactor is then charged with 20.2 Kg 6.0N HCl (aq) solution, the temperature is adjusted to 35° C., and agitation is allowed for at least 12 hours to permit the Boc deprotection reaction to occur. The reaction is monitored by HPLC or reverse phase TLC. When complete, agitation is discontinued and the phases are allowed to separate. The lower, organic phase is removed and set aside. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The organic extracts are combined and discarded. The remaining aqueous phase is diluted with 18.3 Kg distilled water and the temperature is raised to approximately 50° C. Distillation under vacuum (100-400 torr) is performed to remove residual dichloromethane from the reaction. The pH of the reaction is then adjusted to between 7.8-8.1 using about 9.42 Kg of 3.0N NaOH (aq) solution while keeping the temperature of the reaction below 65° C. The reaction is cooled to 50° C. and the precipitated solids are aged for at least an hour prior to cooling the mixture to room temperature. The solids are isolated by suction filtration and washed twice with 5.2 Kg portions of distilled water. The solids are dried for at least 12 hours with suction and then for an additional 12 hours in a convection oven at 55° C. The yield achieved for intermediate (23) in this example is 3.2 Kg (79%). A reactor is charged with 3.2 Kg solid intermediate (23) and the solids are suspended in 25.6 Kg of 95% ethanol as solvent. To the reactor is then added 1.1 Kg of solid D,L-malic acid (24), and the mixture is heated to reflux temperature (˜80° C.). Distilled water (˜5.7 L) is added to the reaction until a complete solution is achieved and 0.2 Kg of activated charcoal is added. The reaction mixture is passed through a filter to achieve clarification, cooled to 45° C. and held for a period of at least 2 hours to allow crystallization to occur. The reaction mixture is further cooled to 5° C. and the suspended solids are isolated by suction filtration. The solids are then washed with 6.6 KG of 95% ethanol and dried for at least 4 hours with suction under vacuum. The solids are then further dried in a convection oven for at least 12 hours at 45° C. to afford 3.1 Kg of intermediate (24) (70%). NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, 1H), 7.37 (d, J=9.0 Hz, 1H), 7.05 (d, J=9.0 Hz, 1H), 4.23-4.18 (m, 1H), 4.10-3.89 (m, 1H), 3.66 (br s, 1H), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, 1H), 3.34 (d, J=9.3 Hz, 1H), 3.16 (d, J=12.9 Hz, 1H), 2.65 (dd, J=16.1, 4.1 Hz, 1H), 2.64-2.53 (m, 1H), 2.46 (dd, J=16.1, 8.0 Hz, 1H), 2.06 (br s, 1H), 1.87 (d, J=14.4 Hz, 1H), 1.58-1.45 (m, 1H), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H); 0.85-0.78 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization. HPLC: Mobile Phase H2O with 0.1% formic acid/Acetonitrile with 0.1% formic acid, gradient elution with 88% H2O/formic acid to 20% H2O/formic acid, Zorbax SB-C8 4.6 mm×150 mm column, Part No. 883975.906, 1.5 ml/min rate, 20 min run time, 292 nm, Detector Model G1314A, S/N JP72003849, Quat Pump Model G1311A, S/N US72102299, Auto Sampler Model G1313A, S/N DE14918139, Degasser Model G1322A, S/N JP73007229; approximate retention time for intermediate (19): 13.0 min; approximate retention time for intermediate (20): 11.6 min; approximate retention time for intermediate (21): 16.3 min; approximate retention time for intermediate (22): 18.2 min; approximate retention time for intermediate (23): 8.6 min; approximate retention time for compound (25): 8.6 min.
....................
REF
A. ARJONA ET AL: "Nemonoxacin", DRUGS OF THE FUTURE, vol. 34, no. 3, 1 January 2009 (2009-01-01), page 196, XP55014485, ISSN: 0377-8282, DOI: 10.1358/dof.2009.034.03.1350294
| 2 | * | ANONYMOUS: "TaiGen Announces Positive Data From the Phase II Study of Nemonoxacin (TG-873870) in Community-Acquired Pneumonia", INTERNET CITATION, [Online] 7 April 2008 (2008-04-07), page 1, XP007919900, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#16> [retrieved on 2011-12-12] |
| 3 | * | ANONYMOUS: "TaiGen Biotechnology Initiates Phase II Trial Of Nemonoxacin For Treatment Of Adult Community Acquired Pneumonia (CAP)", 20070108, [Online] 8 January 2007 (2007-01-08), page 1, XP007919910, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#11> [retrieved on 2011-12-12] |
| 4 | * | ANONYMOUS: "TaiGen Initiates Phase 1B Trial of a Novel Quinolone Antibiotic", 20050618, 18 June 2005 (2005-06-18), pages 1-2, XP007919904, |
| 5 | * | See also references of WO2010002415A1 |
| WO2007110834A2 * | Mar 26, 2007 | Oct 4, 2007 | Procter & Gamble | Malate salts, and polymorphs of (3s,5s)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid |
| WO2009023473A2 * | Aug 5, 2008 | Feb 19, 2009 | Chi-Hsin Richard King | Antimicrobial parenteral formulation |
| WO2010009014A2 * | Jul 10, 2009 | Jan 21, 2010 | Taigen Biotechnology Co., Ltd. |
| US8211909 | 7-4-2012 | TREATMENT OF ANTIBIOTIC-RESISTANT BACTERIA INFECTION |
| US8158798 | 4-18-2012 | Coupling Process For Preparing Quinolone Intermediates |
| US8039485 | 10-19-2011 | Malate salts, and polymorphs of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid |
| US2010152452 | 6-18-2010 | STEREOSELECTIVE SYNTHESIS OF PIPERIDINE DERIVATIVES |
| US2010041697 | 2-19-2010 | PNEUMONIA TREATMENT |
| US7528264 | 5-6-2009 | Hydride reduction process for preparing quinolone intermediates |
| US2009042932 | 2-13-2009 | ANTIMICROBIAL PARENTERAL FORMULATION |
| US7456279 | 11-26-2008 | Coupling process for preparing quinolone intermediates |
| US8158798 | Oct 27, 2008 | Apr 17, 2012 | Taigen Biotechnology Co., Ltd. | Coupling process for preparing quinolone intermediates |
| US8211909 | Sep 8, 2008 | Jul 3, 2012 | Taigen Biotechnology Co., Ltd. | Treatment of antibiotic-resistant bacteria infection |
| WO2010002965A2 * | Jul 1, 2009 | Jan 7, 2010 | Taigen Biotechnology Co., Ltd. | Pneumonia treatmen |
WO 2007110834
WO 2007110835
WO 2007110836
WO 1999014214
WO 2010077798

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.
No comments:
Post a Comment