
> Asfotase Alfa Sequence LVPEKEKDPKYWRDQAQETLKYALELQKLNTNVAKNVIMFLGDGMGVSTVTAARILKGQL HHNPGEETRLEMDKFPFVALSKTYNTNAQVPDSAGTATAYLCGVKANEGTVGVSAATERS RCNTTQGNEVTSILRWAKDAGKSVGIVTTTRVNHATPSAAYAHSADRDWYSDNEMPPEAL SQGCKDIAYQLMHNIRDIDVIMGGGRKYMYPKNKTDVEYESDEKARGTRLDGLDLVDTWK SFKPRYKHSHFIWNRTELLTLDPHNVDYLLGLFEPGDMQYELNRNNVTDPSLSEMVVVAI QILRKNPKGFFLLVEGGRIDHGHHEGKAKQALHEAVEMDRAIGQAGSLTSSEDTLTVVTA DHSHVFTFGGYTPRGNSIFGLAPMLSDTDKKPFTAILYGNGPGYKVVGGERENVSMVDYA HNNYQAQSAVPLRHETHGGEDVAVFSKGPMAHLLHGVHEQNYVPHVMAYAACIGANLGHC APASSLKDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEV KFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIE KTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKT TPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKDIDDDD DDDDDD
Asfotase alfa
Indicated for the treatment of patients with perinatal/infantile and juvenile onset hypophosphatasia (HPP).
(Strensiq®)Approved
A mineralized tissue targeted fusion protein used to treat hypophosphatasia.
Research Code ALXN-1215; ENB-0040; sALP-FcD-10
CAS No.1174277-80-5
| 180000.0 C7108H11008N1968O2206S56 |

| Company | Alexion Pharmaceuticals Inc. |
| Description | Fusion protein incorporating the catalytic domain of human tissue non-specific alkaline phosphatase (TNSALP; ALPL) and a bone-targeting peptide |
| Molecular Target | |
| Mechanism of Action | Enzyme replacement therapy |
| Therapeutic Modality | Biologic: Fusion protein |
| Latest Stage of Development | Approved |
| Standard Indication | Metabolic (unspecified) |
| Indication Details | Treat hypophosphatasia (HPP); Treat hypophosphatasia (HPP) in children; Treat hypophosphatasia (HPP) in patients whose first signs or symptoms occurred prior to 18 years of age; Treat perinatal, infantile and juvenile-onset hypophosphatasia (HPP) |
| Regulatory Designation | U.S. - Breakthrough Therapy (Treat hypophosphatasia (HPP) in children); U.S. - Breakthrough Therapy (Treat hypophosphatasia (HPP) in patients whose first signs or symptoms occurred prior to 18 years of age); U.S. - Fast Track (Treat hypophosphatasia (HPP)); U.S. - Orphan Drug (Treat hypophosphatasia (HPP)); U.S. - Priority Review (Treat hypophosphatasia (HPP) in children); EU - Accelerated Assessment (Treat hypophosphatasia (HPP)); EU - Accelerated Assessment (Treat hypophosphatasia (HPP) in children); EU - Orphan Drug (Treat hypophosphatasia (HPP)); Japan - Orphan Drug (Treat hypophosphatasia (HPP)); Australia - Orphan Drug (Treat hypophosphatasia (HPP) |
Hypophosphatasia (HPP) is a rare inheritable disease that results from loss-of-function mutations in the ALPL gene encoding tissue-nonspecific alkaline phosphatase (TNSALP). Therapeutic options for treating the underlying pathophysiology of the disease have been lacking, with the mainstay of treatment being management of symptoms and supportive care. HPP is associated with significant morbidity and mortality in paediatric patients, with mortality rates as high as 100 % in perinatal-onset HPP and 50 % in infantile-onset HPP. Subcutaneous asfotase alfa (Strensiq(®)), a first-in-class bone-targeted human recombinant TNSALP replacement therapy, is approved in the EU for long-term therapy in patients with paediatric-onset HPP to treat bone manifestations of the disease. In noncomparative clinical trials in infants and children with paediatric-onset HPP, asfotase alfa rapidly improved radiographically-assessed rickets severity scores at 24 weeks (primary timepoint) as reflected in improvements in bone mineralization, with these benefits sustained after more than 3 years of treatment. Furthermore, patients typically experienced improvements in respiratory function, gross motor function, fine motor function, cognitive development, muscle strength (normalization) and ability to perform activities of daily living, and catch-up height-gain. In life-threatening perinatal and infantile HPP, asfotase alfa also improved overall survival. Asfotase alfa was generally well tolerated in clinical trials, with relatively few patients discontinuing treatment and most treatment-related adverse events being of mild to moderate intensity. Thus, subcutaneous asfotase alfa is a valuable emerging therapy for the treatment of bone manifestations in patients with paediatric-onset HPP.
FDA
October 23, 2015
Release
Today, the U.S. Food and Drug Administration approved Strensiq (asfotase alfa) as the first approved treatment for perinatal, infantile and juvenile-onset hypophosphatasia (HPP).
HPP is a rare, genetic, progressive, metabolic disease in which patients experience devastating effects on multiple systems of the body, leading to severe disability and life-threatening complications. It is characterized by defective bone mineralization that can lead to rickets and softening of the bones that result in skeletal abnormalities. It can also cause complications such as profound muscle weakness with loss of mobility, seizures, pain, respiratory failure and premature death. Severe forms of HPP affect an estimated one in 100,000 newborns, but milder cases, such as those that appear in childhood or adulthood, may occur more frequently.
“For the first time, the HPP community will have access to an approved therapy for this rare disease,” said Amy G. Egan, M.D., M.P.H., deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research (CDER). “Strensiq’s approval is an example of how the Breakthrough Therapy Designation program can bring new and needed treatments to people with rare diseases.”
Strensiq received a breakthrough therapy designation as it is the first and only treatment for perinatal, infantile and juvenile-onset HPP. The Breakthrough Therapy Designation program encourages the FDA to work collaboratively with sponsors, by providing timely advice and interactive communications, to help expedite the development and review of important new drugs for serious or life-threatening conditions. In addition to designation as a breakthrough therapy, the FDA granted Strensiq orphan drug designation because it treats a disease affecting fewer than 200,000 patients in the United States.
Orphan drug designation provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Strensiq was also granted priority review, which is granted to drug applications that show a significant improvement in safety or effectiveness in the treatment of a serious condition. In addition, the manufacturer of Strensiq was granted a rare pediatric disease priority review voucher – a provision intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. Development of this drug was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and/or effectiveness of products for use in rare diseases or conditions.
Strensiq is administered via injection three or six times per week. Strensiq works by replacing the enzyme (known as tissue-nonspecific alkaline phosphatase) responsible for formation of an essential mineral in normal bone, which has been shown to improve patient outcomes.
The safety and efficacy of Strensiq were established in 99 patients with perinatal (disease occurs in utero and is evident at birth), infantile- or juvenile-onset HPP who received treatment for up to 6.5 years during four prospective, open-label studies. Study results showed that patients with perinatal- and infantile-onset HPP treated with Strensiq had improved overall survival and survival without the need for a ventilator (ventilator-free survival). Ninety-seven percent of treated patients were alive at one year of age compared to 42 percent of control patients selected from a natural history study group. Similarly, the ventilator-free survival rate at one year of age was 85 percent for treated patients compared to less than 50 percent for the natural history control patients.
Patients with juvenile-onset HPP treated with Strensiq showed improvements in growth and bone health compared to control patients selected from a natural history database. All treated patients had improvement in low weight or short stature or maintained normal height and weight. In comparison, approximately 20 percent of control patients had growth delays over time, with shifts in height or weight from the normal range for children their age to heights and weights well below normal for age. Juvenile-onset patients also showed improvements in bone mineralization, as measured on a scale that evaluates the severity of rickets and other HPP-related skeletal abnormalities based on x-ray images. All treated patients demonstrated substantial healing of rickets on x-rays while some natural history control patients showed increasing signs of rickets over time.
The most common side effects in patients treated with Strensiq include injection site reactions, hypersensitivity reactions (such as difficulty breathing, nausea, dizziness and fever), lipodystrophy (a loss of fat tissue resulting in an indentation in the skin or a thickening of fat tissue resulting in a lump under the skin) at the injection site, and ectopic calcifications of the eyes and kidney.
Strensiq is manufactured by Alexion Pharmaceuticals Inc., based in Cheshire, Connecticut.
HPP is a rare, genetic, progressive, metabolic disease in which patients experience devastating effects on multiple systems of the body, leading to severe disability and life-threatening complications. It is characterized by defective bone mineralization that can lead to rickets and softening of the bones that result in skeletal abnormalities. It can also cause complications such as profound muscle weakness with loss of mobility, seizures, pain, respiratory failure and premature death. Severe forms of HPP affect an estimated one in 100,000 newborns, but milder cases, such as those that appear in childhood or adulthood, may occur more frequently.
“For the first time, the HPP community will have access to an approved therapy for this rare disease,” said Amy G. Egan, M.D., M.P.H., deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research (CDER). “Strensiq’s approval is an example of how the Breakthrough Therapy Designation program can bring new and needed treatments to people with rare diseases.”
Strensiq received a breakthrough therapy designation as it is the first and only treatment for perinatal, infantile and juvenile-onset HPP. The Breakthrough Therapy Designation program encourages the FDA to work collaboratively with sponsors, by providing timely advice and interactive communications, to help expedite the development and review of important new drugs for serious or life-threatening conditions. In addition to designation as a breakthrough therapy, the FDA granted Strensiq orphan drug designation because it treats a disease affecting fewer than 200,000 patients in the United States.
Orphan drug designation provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Strensiq was also granted priority review, which is granted to drug applications that show a significant improvement in safety or effectiveness in the treatment of a serious condition. In addition, the manufacturer of Strensiq was granted a rare pediatric disease priority review voucher – a provision intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. Development of this drug was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and/or effectiveness of products for use in rare diseases or conditions.
Strensiq is administered via injection three or six times per week. Strensiq works by replacing the enzyme (known as tissue-nonspecific alkaline phosphatase) responsible for formation of an essential mineral in normal bone, which has been shown to improve patient outcomes.
The safety and efficacy of Strensiq were established in 99 patients with perinatal (disease occurs in utero and is evident at birth), infantile- or juvenile-onset HPP who received treatment for up to 6.5 years during four prospective, open-label studies. Study results showed that patients with perinatal- and infantile-onset HPP treated with Strensiq had improved overall survival and survival without the need for a ventilator (ventilator-free survival). Ninety-seven percent of treated patients were alive at one year of age compared to 42 percent of control patients selected from a natural history study group. Similarly, the ventilator-free survival rate at one year of age was 85 percent for treated patients compared to less than 50 percent for the natural history control patients.
Patients with juvenile-onset HPP treated with Strensiq showed improvements in growth and bone health compared to control patients selected from a natural history database. All treated patients had improvement in low weight or short stature or maintained normal height and weight. In comparison, approximately 20 percent of control patients had growth delays over time, with shifts in height or weight from the normal range for children their age to heights and weights well below normal for age. Juvenile-onset patients also showed improvements in bone mineralization, as measured on a scale that evaluates the severity of rickets and other HPP-related skeletal abnormalities based on x-ray images. All treated patients demonstrated substantial healing of rickets on x-rays while some natural history control patients showed increasing signs of rickets over time.
The most common side effects in patients treated with Strensiq include injection site reactions, hypersensitivity reactions (such as difficulty breathing, nausea, dizziness and fever), lipodystrophy (a loss of fat tissue resulting in an indentation in the skin or a thickening of fat tissue resulting in a lump under the skin) at the injection site, and ectopic calcifications of the eyes and kidney.
Strensiq is manufactured by Alexion Pharmaceuticals Inc., based in Cheshire, Connecticut.

| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US7763712 | No | 2004-04-21 | 2026-07-15 |
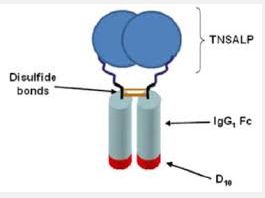
STRENSIQ is a formulation of asfotase alfa, which is a soluble glycoproteincomposed of two identical polypeptide chains. Each chain contains 726amino acids with a theoretical mass of 161 kDa. Each chain consists of the catalytic domain of human tissue non-specific alkaline phosphatase (TNSALP), the human immunoglobulin G1 Fc domain and a deca-aspartatepeptide used as a bone targeting domain. The two polypeptide chains are covalently linked by two disulfide bonds.
STRENSIQ is a tissue nonspecific alkaline phosphatase produced byrecombinant DNA technology in a Chinese hamster ovary cell line. TNSALP is a metallo-enzyme that catalyzes the hydrolysis of phosphomonoesters with release of inorganic phosphate and alcohol. Asfotase alfa has a specific activity of 620 to 1250 units/mg. One activity unit is defined as the amount of asfotase alfa required to form 1 μmol of p-nitrophenol from pNPP per minute at 37°C.
STRENSIQ (asfotase alfa) is a sterile, preservative-free, nonpyrogenic, clear, slightly opalescent or opalescent, colorless to slightly yellow, with few small translucent or white particles, aqueous solution for subcutaneous administration. STRENSIQ is supplied in glass single-use vials containing asfotase alfa; dibasic sodium phosphate, heptahydrate; monobasic sodium phosphate, monohydrate; and sodium chloride at a pH between 7.2 and 7.6. Table 5 describes the content of STRENSIQ vial presentations.
Table 5: Content of STRENSIQ Vial Presentations
| INGREDIENT | QUANTITY PER VIAL | |||
| ASFOTASE ALFA | 18 MG/0.45 ML | 28 MG/0.7 ML | 40 MG/ML | 80 MG/0.8 ML |
| Dibasic sodium phosphate, heptahydrate | 2.48 mg | 3.85 mg | 5.5 mg | 4.4 mg |
| Monobasic sodium phosphate, monohydrate | 0.28 mg | 0.43 mg | 0.62 mg | 0.5 mg |
| Sodium chloride | 3.94 mg | 6.13 mg | 8.76 mg | 7.01 mg |
REFERENCES
- Whyte MP: Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016 Apr;12(4):233-46. doi: 10.1038/nrendo.2016.14. Epub 2016 Feb 19. [PubMed:26893260 ]
- Whyte MP, Rockman-Greenberg C, Ozono K, Riese R, Moseley S, Melian A, Thompson DD, Bishop N, Hofmann C: Asfotase Alfa Treatment Improves Survival for Perinatal and Infantile Hypophosphatasia. J Clin Endocrinol Metab. 2016 Jan;101(1):334-42. doi: 10.1210/jc.2015-3462. Epub 2015 Nov 3. [PubMed:26529632 ]
- Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS, Bauer ML, Hamdan MA, Bishop N, Lutz RE, McGinn M, Craig S, Moore JN, Taylor JW, Cleveland RH, Cranley WR, Lim R, Thacher TD, Mayhew JE, Downs M, Millan JL, Skrinar AM, Crine P, Landy H: Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012 Mar 8;366(10):904-13. doi: 10.1056/NEJMoa1106173. [PubMed:22397652 ]
//////Asfotase alfa, Strensiq, treat hypophosphatasia, ALXN-1215, ENB-0040, sALP-FcD-10, FDA 2015
No comments:
Post a Comment