LY-156735 (TIK-301, PD-6735)….for the treatment of sleep latency in patients with primary insomnia
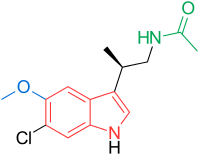
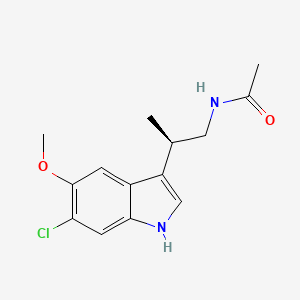
N-[(2R)-2-(6-chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide
LY-156735 (TIK-301, PD-6735) is a melatonin MT1 and MT2 agonist which is under development for the treatment of insomnia and other sleep disorders.[1]
Beta-methyl-6-chloromelatonin (PD-6735) is a melatonin MT1 and MT2 agonist which had been in phase II trials at Phase 2 Discovery for the treatment of sleep latency in patients with primary insomnia, however, no recent development has been reported.
The melatonin agonist exhibits high selectivity and provides a novel mode of action different from that of benzodiazepine receptor ligands currently on the market.
Furthermore, the drug candidate is believed to be non-addicting, therefore, offering an advantage over marketed sleep medications. Originally discovered by Lilly, PD-6735 was licensed to Phase 2 Discovery in 2002 for further development.
Orphan drug designation has been assigned in the U.S. for the treatment of circadian rhythm sleep disorders in blind people with no light perception and for the treatment of neuroleptic-induced tardive dyskinesia in schizophrenia patients.
In 2007, the product candidate was licensed to Tikvah Therapeutics by Phase 2 Discovery for worldwide development and commercialization for the treatment of sleep disorder, depression and circadian rhythm disorder.
beta -alkylmelatonins as ovulation inhibitors [US4997845]1991-03-05
BETA-ALKYLMELATONINS [EP0281242]1988-09-07 GRANT1992-08-12

The condensation of 6-chloro-5-methoxy-1H-indole (I) with Meldrum’s acid (II) and acetaldehyde (III) catalyzed by L-proline in acetonitrile gives the adduct (IV), which is treated with Cu and ethanol in refluxing pyridine to yield 3-(6-chloro-5-methoxy-1H-indol-3-yl)butyric acid ethyl ester (V). The reaction of (V) with hydrazine at 140 C affords the hydrazide (VI), which is treated with NaNO2 and Ac-OH to provide the corresponding azide that, without isolation, is thermolyzed and rearranged in toluene at 80?C to give 7-chloro-6-methoxy-4-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (VII). The cleavage of the lactam ring of (VII) with KOH in refluxing ethanol/water yields 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (VIII). The decarboxylation of (VIII) by means of refluxing aq. 3M HCl affords 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole (IX), which is finally acylated with acetic anhydride and pyridine in toluene to provide the target 6-chloromelatonin as a racemic compound.
EP 0281242;……….http://www.google.com/patents/EP0281242B1?cl=en
Example 3
- Preparation of β-Methyl-6-chloromelatonin
- Following the procedure of Example 1, a solution of 10.0 g (0.055 mole) of 5-methoxy-6-chloroindole, 3.1 ml (2.44 g, 0.055 mole) of acetaldehyde, and 7.94 g (0.055 mole) of Meldrum’s acid in 90 ml of acetonitrile was stirred for 48 hours. The solvent was removed under vacuum, and the adduct thus prepared was recrystallized by dissolving in warm toluene and immediately cooling. The adduct was obtained as slightly pink crystals; m.p. = 145°C; yield = 16.5 g (85%). The elemental analysis of the product showed a slightly elevated percentage of carbon. However, the NMR spectrum indicated that the product was pure and had the indicated structure.
Analysis calc. for C₁₇H₁₈NO₅Cl- Theory:
- C, 58.04; H, 5.16; N, 3.98; Cl, 10.08
- Found :
- C, 59.34; H, 5.15; N, 3.84; Cl, 9.69
- The solvolysis and decarboxylation of the adduct (11.0 g; 31.3 mmoles) using ethanol, pyridine, and copper dust was carried out by the procedure of Example 1. The yield of 3-(5-methoxy-6-chloro-1H-indol-3-yl)pentanoic acid ethyl ester, a pale yellow oil, after chromatography over silica gel using 10% EtOAc/90% toluene was 8.68 g (94%).
Analysis calc. for C₁₅H₁₈NO₃Cl- Theory:
- C, 60.91; H, 6.13; N, 4.74; Cl, 11.99
- Found :
- C, 60.67; H, 5.86; N, 4.93; Cl, 11.73
- A mixture of 8.68 g (29.3 mmoles) of the above ethyl ester and 6 ml of hydrazine hydrate was heated at 140°C under nitrogen in a flask fitted with an air cooled condensor. After 6½ hours, the excess hydrazine hydrate was removed under vacuum. The 2-methyl-2-(5-methoxy-6-chloro-3-indolyl)-propionhydrazide thus prepared was recrystallized from ethyl acetate; Yield = 7.13 g (86%); m.p. = 154-155°C.
Analysis calc. for C₁₃H₁₆N₃O₂Cl- Theory:
- C, 55.42; H, 5.72; N, 14.91; Cl, 12.58
- Found :
- C, 55.14; H, 5.51; N, 14.49; Cl, 12.78
- The above hydrazide (7.13 g, 25 mmoles) was converted to the corresponding acyl azide, the azide thermolyzed and rearranged at 80° in toluene, and the rearranged product cyclized with HCl according to the procedure of Example 1. The yield of crude, light tan, lactam, 1-oxo-4-methyl-6-methoxy-7-chloro-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indole, product, (m.p. = 249-252°C) was 4.77 g (72%).
Analysis calc. for C₁₃H₁₃N₂O₂Cl- Theory:
- C, 58.99; H, 4.95; N, 10.58
- Found :
- C, 59.45; H, 4.77; N, 10.72
- The crude lactam (4.77 g, 18 mmoles) was hydrolyzed with aqueous ethanolic KOH as described in Example 1. The yield of crude amino acid, 2-carboxy-3-(1-amino-2-propyl)-5-methoxy-6-chloroindole, was 3.98 g (78%). The crude product (3.0 g; 10.6 mmoles) was decarboxylated, using the procedure of Example 1, by refluxing in 100 ml of 3M HCl overnight. The acidic solution was decolorized with activated carbon and was then basified with 5M NaOH. The amine was extracted into diethyl ether. After drying the ether extract over Na₂SO₄, the diethyl ether was removed in vacuoleaving as a residue the crystallized tryptamine, 3-(1-amino-2-propyl)-5-methoxy-6-chloroindole; m.p. 133-4°C. The yield, after recrystallization from toluene/hexane, was 1.62 g (64%).
Analysis calc. for C₁₂H₁₅N₂OCl- Theory:
- C, 60.38; H, 6.33; N, 11.74; Cl, 14.85
- Found :
- C, 60.11; H, 6.05; N, 11.93; Cl, 15.06
- A solution of 1.51 g (6.3 mmoles) of the above tryptamine in 10 ml of toluene and 2.5 ml of pyridine was treated with 1.5 ml of acetic anhydride. After allowing the reaction mixture to stand for three hours at room temperature, the volatile materials were removed under vacuum. The residue was dissolved in ethyl acetate, and washed with aqueous NaHCO₃, and brine. The ethyl acetate solution was dried over Na₂SO₄, and the solvent removed by evaporation. The residual oil was crystallized from toluene/hexane yielding 6-chloro-β-methylmelatonin, (m.p. = 133-5°C; 1.09 g, 61%).
Analysis calc. for C₁₄H₁₇N₂O₂Cl- Theory:
- C, 59.89; H, 6.10; N, 9.98; Cl, 12.63
- Found :
- C, 60.03; H, 6.22; N, 9.75; Cl, 12.92
…………………………………………….
PATENT

The intermediate diazonium salt (XIII) has been obtained as follows: the hydrogenation of 3-chloro-4-methoxynitrobenzene (XI) with H2 over Pt/Al2O3 in toluene gives the corresponding aniline (XII), which is diazotized with NaNO2/HCl and treated with sodium tetrafluoroborate to yield the target diazonium salt intermediate (XIII). The reduction of pulegone (I) with H2 over Pd/C gives the menthol (II), which is oxidized with CrO3/H2SO4 to yield 3(R),7-dimethyl-6-oxooctanoic acid (IV), which can also be obtained by direct oxidation of (l)-menthol (III) under the same conditions.
The oxidation of (IV) with trifluoroperacetic acid (trifluoroacetic anhydride/H2O2) in dichloromethane yields the 3(R)-methylhexanedioic acid isopropyl monoester (V), which is treated with NaOEt in ethanol to obtain the corresponding ethyl monoester (VI). The reaction of (VI) with diethyl carbonate, EtONa, and “Adogen 464″ (a phase transfer catalyst) in ethanol affords 5,5-bis(ethoxycarbonyl)-3(S)-methylpentanoic acid (VII), which is treated with oxalyl chloride to provide the expected acyl chloride (VIII). The reaction of (VIII) with sodium azide and benzyl alcohol gives the intermediate azide that rearranges to the benzyl carbamate (IX).
The reductive cyclization of (IX) with H2 over Pd/C in ethanol yields 5(R)-methyl-2-oxopiperidine-3-carboxylic acid ethyl ester (X), which is condensed with the intermediate diazonium salt (XIII) to afford the hydrazono derivative (XIV). The cyclization of (XIV) in hot formic acid provides 7-chloro-6-methoxy-4(R)-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (XV), which is treated with KOH In refluxing ethanol/water to cleave the lactam ring, yielding 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (XVI). The decarboxylation of (XVI) by means of refluxing 3M HCl affords 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole (XVII), which is finally acylated with Ac2O and pyridine in toluene to provide the target 6-chloromelatonin as a pure enantiomer.
Example 7
- Preparation of S-(-)-β-methyl-6-chloromelatonin and R-(+)-β-methyl-6-chloromelatonin
- A solution of 4.0 g (21 mmoles) of 3-chloro-4-methoxynitrobenzene in 200 ml of toluene was hydrogenated over 0.4 g of 5% platinum on alumina. The catalyst was removed by filtration and the solvent evaporated from the filtrate. The crude 3-chloroanisidine prepared was placed in solution in diethyl ether and treated with ethereal HCl to produce the hydrochloride salt, which was collected and dried; weight = 2.48 g (61% yield).
- A mixture of 2.40 g (12.4 mmoles) of 3-chloroanisidine hydrochloride in 7 ml of 4M HCl was treated, at 0°C, with 0.86 g (12.5 mmoles) of sodium nitrite in 5 ml of water. After stirring at 0°C for an hour the solution was filtered and the filtrate added slowly to an ice cold solution of 2.6 g (24 mmoles) of sodium fluoroborate in 8 ml of water. After stirring at 0°C for an hour the salt was collected and washed successively with, cold 5% sodium fluoroborate solution, cold methanol, and ether. The dried 3-chloro-4-methoxybenzene diazonium fluoroborate thus prepared weighed 2.2 g (69% yield).
- A mixture of 2.03 g (11.0 mmole) of (R)-(-)-3-ethoxycarbonyl-5-methyl-2-piperidone and 30 ml of 0.75M NaOH was stirred at room temperature (24°C) overnight. The solution was cooled to 0°C and the pH lowered to 3.5 with 3M hydrochloric acid. The diazonium salt (2.8 g, 10.9 mmoles) was added in small portions and the reaction mixture cooled to about 0°C overnight. The product, R-(-)-3-(3-chloro-4-methoxy)phenylhydrazono-5-methyl-2-piperidone, was collected, washed with water, and dried; weight = 2.30 g (75% yield); m.p. = 205°C. A small sample was further purified by chromatography over a short silica gel column using ethyl acetate as the eluant. [α]²⁵ = -58° (c = 10, MeOH).
Analysis calc. for C₁₃H₁₆N₃O₂Cl- Theory:
- C, 55.42; H, 5.72; N, 14.91; Cl, 12.58
- Found :
- C, 55.79; H, 5.78: N, 14.72; Cl, 12.69
- A mixture of 2.20 g (7.8 moles) of the R-(-) hydrazone and 20 ml of 90% formic acid was heated at 85° for three hours then slowly diluted with an equal volume of water. The mixture was allowed to cool and then chilled overnight. The dark precipitate was collected, washed with water, then recrystallized from acetone/water, yielding 1.20 g (60% yield) of S-(-)-1-oxo-4-methyl-6-methoxy-7-chloro-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indole; m.p. = 248°C. [α]²⁵ = -12.2° (c = 10, MeOH).
Analysis calc. for C₁₃H₁₃N₂O₂Cl- Theory:
- C, 58.99; H, 4.95; N, 10.58; Cl, 13.39
- Found :
- C, 59.16; H, 4.88; N, 10.80; Cl, 13.15
- The conversion of (S)-(-)-lactam to (S)-(-)-6-chloro-β-methylmelatonin was carried out as described previously in Example 3. The product, S-(-)-β-methyl-6-chloromelatonin, was spectroscopically identical to the racemate, but gave an optical rotation of [α]²⁵ = -13.2° (c = 10, MeOH).
- (R)-(+)-6-chloro-β-methylmelatonin was synthesized from (S)-(+)-3-ethoxycarbonyl-5-methyl-2-piperidone in the same manner as described above. The stereoisomer was identical to the (S)-(-) material except for the sign of rotation.
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| N-[(2R)-(6-Chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide | |
| CLINICAL DATA | |
| LEGAL STATUS |
?
|
| IDENTIFIERS | |
| CAS NUMBER | 118702-11-7 |
| ATC CODE | ? |
| PUBCHEM | CID 219018 |
| CHEMSPIDER | 189853 |
| CHEMICAL DATA | |
| FORMULA | C14H17ClN2O2 |
| MOLECULAR MASS | 280.757 |
























