334826-98-1 free form
334827-98-4 (as besylate)
334827-98-4 (as besylate)
- UK 369003
- UK-369,003
- UK0369,003
- UNII-S6G4R7DI1C
THERAPEUTIC CLAIM Treatment of lower urinary tract
symptoms associated with BPH
symptoms associated with BPH
CHEMICAL NAMES FREE FORM
1. ........7H-Pyrazolo[4,3-d]pyrimidin-7-one, 5-[2-ethoxy-5-[(4-ethyl-1-
piperazinyl)sulfonyl]-3-pyridinyl]-3-ethyl-2,6-dihydro-2-(2-methoxyethyl)-
piperazinyl)sulfonyl]-3-pyridinyl]-3-ethyl-2,6-dihydro-2-(2-methoxyethyl)-
2. .......5-{2-ethoxy-5-[(4-ethylpiperazin-1-yl)sulfonyl]pyridin-3-yl}-3-ethyl-2-(2-
methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one
methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one
3.........1-(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine
MOLECULAR FORMULA C23H33N7O5S
MOLECULAR WEIGHT 519.6
CODE DESIGNATION UK-369,003
CAS REGISTRY NUMBER 334826-98-1
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulfonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-6,7-dihydro-2H-pyrazolo[4,3-d]pyrimidin-7-one
Phosphodiesterase PDE5A Inhibitors , Treatment of Erectile Dysfunction
Pfizer (Originator)
UK-369003 is a phosphodiesterase V (PDE V) inhibitor which had been under development for the treatment of erectile dysfunction, pulmonary hypertension and for the treatment of lower urinary tract symptoms, but no recent development has been reported for these indications. Trials for the treatment of benign prostatic hyperplasia were discontinued.
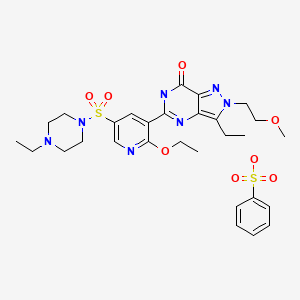 Gisadenafil besylate (USAN)
Gisadenafil besylate (USAN)
D09622, 334827-98-4
M.Wt:677.79
5-(2-ethoxy-5-(4-ethylpiperazin-1-ylsulfonyl)pyridin-3-yl)-3-ethyl-2-(2-methoxyethyl)-2H-pyrazolo[4,3-d]pyrimidin-7(6H)-one benzenesulfonate
1-[[6-Ethoxy-5-[3-ethyl-4,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridinyl]sulfonyl]-4-ethylpiperazine Monobenzenesulfonate
Formula:C23H33N7O5S.C6H6O3S
| Certificate of Analysis |
|
| Biological Activity:Potent and selective PDE5 inhibitor (IC50: 1.23 nM) with improved selectivity over PDE6(PDE5/6 selectivity value 117 and >3000-fold selectivity over other PDEs).Gisadenafil has the potential for oral bioavailability and dose-proportional pharmacokinetics. Close analogue of Sildenafil (Viagra; Axon 2046) |
Gisadenafil besylate is a PDE5 inhibitor. Inhibition of PDE5 prevents the breakdown of cyclic phosphodiester secondary messenger molecules. This has the effect of prolonging and enhancing signal transduction.
CLINICAL TRIALS
...............................
PAPERS

Bioorganic and Medicinal Chemistry, 2012 , vol. 20, 1 p. 498 - 509


Scheme 1.
Reagents and conditions: (i) 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, hydroxybenzotriazole, di-isopropylethylamine, THF, 20 °C, 20 h; (ii) caesium carbonate, alkyl mesylate or alkyl chloride, DMF, 20 °C, 20 h; (iii) KHMDS, R1OH, 120 °C, 20 h.

Scheme 2.
Reagents and conditions: (i) KHMDS, nBuOH, 120–130 °C, pressure vessel (ii) TFA, CH2Cl2; (iii) methanesulphonyl chloride, NEt3, CH2Cl2; (iv) HOAc, NaCNBH3, CH2O (v) KHMDS, nBuOH, reflux.

Scheme 3.
Reagents and conditions: (i) caesium carbonate, RCl, DMF; (ii) 50 psi H2, 10% Pd/C (iii) 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, HOBT, di-isopropylethylamine, THF, 20 °C, 20 h; (iv) KHMDS, ethanol, 120 °C, pressure vessel; (v) TFA, CH2Cl2; (vi) CH2O, HOAc, NaCNBH3; (vii) R1OH, KHMDS, 120 °C.

Scheme 4.
Reagents and conditions: (i) NaNO2, HCl, H2O; (ii) TFAA, Et2O; (iii) ethyl propynoate, xylene, reflux, 2 h; (iv) NaOH, H2O, dioxan; (v) HNO3/H2SO4, 40–55 °C; (vi) (COCl)2, CH2Cl2, DMF; (vii) NH3, THF; (viii) 10% Pd/C, EtOH, 60 psi H2, 20 °C, 14 h; (ix) acid chloride of 3, NEt3, CH2Cl2; (x) KHMDS, EtOH, 130 °C, 14 h, pressure vessel; (xi) methoxyethanol, KHMDS, reflux, 14 h.
.................................
PAPERS
Org. Proc. Res. Dev., 2004, 8 (4), pp 674–679
DOI: 10.1021/op0300241

...............................
PAPERS
Yousef Hajikarimian, Steve Yeo, Robert W. Ryan, Philip Levett, Christopher Stoneley and Paul Singh
Org Process Res Dev 2010, 14(4): pp 1027–1031
Publication Date (Web): June 25, 2010 (Article)
DOI: 10.1021/op100141g

UK-369,003 was nominated for development as the lead candidate for treatment of benign prostatic hyperplasia (BPH). The free base was found to be moderately crystalline with a melting point of 168 °C. Solubility of the free base at physiological pH was found to be poor hence necessitating a comprehensive screen for a suitable salt form of the API. Benzenesulfonic acid was found to form the most suitable counterion for the API with a melting point of 248 °C and satisfied all our requirements for primary and secondary processing. The process for the formation of the benzenesulfonic acid salt involved the use of water/methyl ethyl ketone (4% water by volume) as the reaction medium. The water level at 4% ensured an optimum balance between product quality (purging of impurities) and the reaction yield. The cyclisation reaction (step 2/Scheme 01) involves the use of ethanol as the reaction media. Any residual amount of ethanol in the isolated step 2 product was therefore considered to be a considerable risk factor in the potential formation of ethyl besylate during the final step processing (step 3/Scheme 01).

Scheme 1. Manufacturing route to UK-369,003-26a
aCDI = carbonyl diimidazole; MEK = methyl ethyl ketone; EtOAc = ethyl acetate; KOtBu = potassium tertiary butoxide; EtOH = ethanol.
........................
SYNTHESIS
Compound 1E is also known as 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, or alternatively as 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridyl sulphonyl}-4-ethylpiperazine (the compound of Example 103 of WO 01/27113 and exemplified hereinafter as Example 1).
Preparation 1
2,2-dimethoxybutane:
Methyl ethyl ketone (672 mL) was charged to a 2 L round bottomed flask and stirred at room temperature before being treated with, trimethylorthoformate (763 mL) and para-toluenesulphonic acid (6.65 g, 0.5 mol %). Over a 15 min period the internal temperature rose to 46° C., so the reaction was cooled to 0° C. for 30 min. The reaction was then stirred at room temperature for 2 h. The reaction was then neutralised by pouring onto sodium carbonate (ca. 750 g) with constant stirring. The resultant slurry was filtered under vacuum and the resultant filtrate was distilled at atmospheric pressure. The fraction boiling in the range 118° C.-124° C. was collected as a colourless liquid, 582 g, 70%.
1H NMR (CDCl3): δ=0.88 (3H, t), 1.24 (3H, s), 1.61 (2H, q), 3.17 (6H, s).
Example 1 N-[3-Carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl]-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide
(a) Ethyl 3-ethyl-1H-pyrazole-5-carboxylate (IIA) from (IlI) and (V)

To a stirred solution of 2,2-dimethoxybutane (10 g, 84.7 mMol) in CH2Cl2 (50 mL) under a nitrogen atmosphere at 0° C. was added pyridine (13.7 mL, 169.5 mMol). The reaction mixture was maintained at 0° C. and a solution of trichloroacetyl chloride (18.9 mL, 169.5 mMol) in CH2Cl2 (35 mL) was added over 1 hour with constant stirring. The yellow-orange solution begins to precipitate a white solid as the reaction progresses. The reaction mixture is allowed to warm to room temperature over 20 h. The reaction mixture was diluted with ethanol (150 mL) and re-cooled to 0° C. before treatment with hydrazine hydrate (8.2 mL, 169.5 mMol) as a solution in ethanol (35 mL) over 30 min. The reaction was heated to 50° C. and solvent was distilled at atmospheric pressure. The temperature was increased until the head temperature reached 78° C. Reflux was maintained for a further 2 h, before cooling to room temperature. The reaction mixture was diluted with water (250 mL) and ethanol was removed by evaporation at reduced pressure. The resultant mixture was extracted with CH2Cl2 (3×200 mL). The combined organics were dried (MgSO4), filtered and evaporated at reduced pressure to afford the title compound as a brown oil, 12.05 g, 85%.
1H NMR (300 MHz, CDCl3): δ=1.20 (3H, t), 1.28 (3H, t), 2.67 (2H, q), 4.29 (2H, q), 6.55 (1H, s), 12.56 (1H, s).
LRMS m/z=167.1 [M-H]+, C8H12N2O2 requires 168.2.
(b) Ethyl 3-ethyl-1H-pyrazole-5-carboxylic acid (IIA) from (IIA) via route 1

Aqueous sodium hydroxide solution (10M; 100 ml, 1.0 mol) was added dropwise to a stirred suspension of the title compound of Example (a) (66.0 g, 0.39 mol) in methanol and the resulting solution heated under reflux for 4 hours. The cool reaction mixture was concentrated under reduced pressure to ca. 200 ml, diluted with water (200 ml) and this mixture washed with toluene (3×100 ml). The resulting aqueous phase was acidified with concentrated hydrochloric acid to pH 4 and the white precipitate collected and dried by suction to provide the title compound (34.1 g). δ (DMSOd6): 1.13 (3H,t), 2.56 (2H,q), 6.42 (1H,s).
(c) 4-Nitro-3-n-propyl-1H-pyrazole-5-carboxylic acid
Fuming sulphuric acid (17.8 ml) was added dropwise to stirred, ice-cooled fuming nitric acid (16.0 ml), the resulting solution heated to 50° C., then 3-n-propyl-1H-pyrazole-5-carboxylic acid (Chem. Pharm. Bull., 1984, 32,1568; 16.4 g, 0.106 mol) added portionwise over 30 minutes whilst maintaining the reaction temperature below 60° C. The resulting solution was heated for 18 hours at 60° C., allowed to cool, then poured onto ice. The white precipitate was collected, washed with water and dried by suction to yield the title compound (15.4 g), m.p. 170-172° C. Found: C, 42.35; H, 4.56; N, 21.07. C7H9N3O4requires C, 42.21; H, 4.55; N, 21.10%. δ (DMSOd6): 0.90 (3H,t), 1.64 (2H,m), 2.83 (2H,m), 14.00 (1 H,s).
(d) 3-Ethyl-4-nitro-1H-pyrazole-5-carboxylic acid (IIA) to (AA) via route 2

Obtained from the title compound of Example (b), by analogy with the process of Example (c), as a brown solid (64%). δ (DMSOd6): 1.18 (3H,t), 2.84 (2H,m), 13.72 (1 H,s).
(e) 4-Nitro-3-n-propyl-1H-pyrazole-5-carboxamide
A solution of the title compound of Example (c) (15.4 g, 0.077 mol) in thionyl chloride (75 ml) was heated under reflux for 3 hours and then the cool reaction mixture evaporated under reduced pressure. The residue was azeotroped with tetrahydrofuran (2×50 ml) and subsequently suspended in tetrahydrofuran (50 ml), then the stirred suspension ice-cooled and treated with gaseous ammonia for 1 hour. Water (50 ml) was added and the resulting mixture evaporated under reduced pressure to give a solid which, after trituration with water and drying by suction, furnished the title compound (14.3 g).
m.p. 197-199° C. Found: C, 42.35; H, 5.07; N, 28.38. C7H10N4O3 requires C, 42.42; H, 5.09; N, 28.27%. δ (DMSOd6): 0.90 (3H,t), 1.68 (2H,m), 2.86 (2H,t), 7.68 (1 H,s), 8.00 (1 H,s).
(f) 3-Ethyl-4-nitro-1H-pyrazole-5-carboxamide BA from AA via route 3

Obtained from the title compound of Example (d), by analogy with Example (e), as a white solid (90%). δ (DMSOd6): 1.17 (3H,t), 2.87 (2H,m), 7.40 (1H,s), 7.60 (1H,s), 7.90 (1H,s). LRMS: m/z 185 (M+l)+.
(g)(i) 5-Ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide CA from BA via route 4

A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (2.5 kg, 13.6 Mol), sodium carbonate (1.8 Kg, 17.0 Mol) and 2-bromoethyl methyl ether (1.98 kg, 14.2 Mol) in THF (22.5 L) and water (2.5 L) was heated under reflux and stirred for 20 hours. The mixture was cooled to ambient temperature and CH2Cl2 (67.5 L) and water (22.5 L) were added. The resultant organic and aqueous layers were separated. The aqueous phase was extracted with CH2Cl2 (22.5 L) and the combined organic solution was distilled under atmospheric pressure and replaced with ethyl acetate (33 L) to a final volume of 17 L. The cooled mixture was granulated at ambient temperature for 2 hours, filtered and washed with ethyl acetate (2.5 L). This afforded 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a white crystalline solid, 2.10 kg, 57%. m.p.=140° C. Found: C, 44.46; H, 5.79; N, 23.01. C9H14N4O4 requires C, 44.63; H, 5.79; N, 23.14%.
δ (CDCl3): 1.18 (3H, t), 2.98 (2H, q), 3.22 (3H, s), 3.77 (2H, t), 4.28 (2H, q), 6.03 (1H, s), 7.36 (1H, s).
LRMS: m/z=243 (M+1)+
(g)(ii) 5-Ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide.
A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (25 g, 0.136 Mol), sodium carbonate (18 g, 0.17 Mol) and sodium iodide (20.4 g, 0.136 Mol) were suspended in ethyl methyl ketone (125 mL) at room temperature. 2-bromoethyl methyl ether (12.8 mL, 0.142 Mol) was added and the mixture was heated to reflux and stirred for 70 hours. The mixture was cooled to ambient temperature and water (250 mL) was added. The resultant slurry was warmed to reflux and held at that temperature for 30 min before cooling to room temperature. The resultant precipitate was granulated at room temperature for 3 h, filtered and vacuum dried to afford 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a yellow crystalline solid 24.3 g, 74%. Data as reported for Example (g)(i).
(h) 4-Amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide (IA) from CA via route 5

A mixture of 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide (20 g, 82.6 mMol) and 5% Pd/C (1 g) in methanol (200 mL) was pressurised at 50psi/25° C. in a sealed vessel and stirred for 15 hours. At the end of the reaction the mixture was filtered through arbocel and the filter cake was washed with methanol. The methanolic solution was distilled at atmospheric pressure and replaced with ethyl acetate to a final volume of 100 mL. The cooled mixture was granulated at ambient temperature for 2 h filtered and washed with ethyl acetate (20 mL) to afford 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide as a white crystalline solid, 15 g, 88%. m.p.=131° C. Found: C, 50.75; H, 7.62; N, 26.38. C9H16N4O2 requires C, 50.94; H, 7.55; N, 26.42%. δ (CDCl3): 1.20 (3H, t), 2.63 (2H, q), 3.32 (3H, s), 3.74 (2H, t), 3.95 (2H, s), 4.15 (2H, t), 5.27 (1H, s), 6.59 (1H, s).
LRMS: m/z=213 (M+1)+
(i) N-[3-Carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl]-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide.

2-ethoxy-5-(4-ethyl-1-piperazinylsulfonyl)nicotinic acid (2.31 kg, 6.73 Mol) was suspended in ethyl acetate (16.2 L) and 1,1-carbonyldimidazole (1.09 kg, 6.73 Mol) was added at room temperature. The reaction mixture was heated at 45° C. for 40 minutes and then the reaction was stirred for a further 40 minutes at reflux. After cooling to ambient temperature 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide (1.5 kg, 7.06 Mol) was added to the cooled mixture, and the reaction stirred for a further 15 hours under reflux. The mixture was cooled filtered and the filter cake was washed with 90% water/10% ethyl acetate, (2 mL /g) to afford N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide as an off white crystalline solid, 3.16 kg, 88%. m.p.=156° C. Found: C, 51.33; H, 6.56; N, 18.36. C23H35N7O6S requires C, 51.40; H, 6.53; N, 18.25%.
δ (CDCl3): 1.04 (3H, t), 1.22 (3H, t), 1.60 (3H, t), 2.44 (2H, q), 2.54 (4H, m), 2.96 (2H, q), 3.12 (4H, m), 3.36 (3H, s), 3.81 (2H, t), 4.27 (2H, t), 4.80(2H, q), 5.35(1H, s), 6.68 (1H, s), 8.66 (1H, d), 8.86 (1H, d), 10.51 (1H, s).
LRMS: m/z=539 (M+1)+
(i) 1-(6-Ethoxy-5-[3-ethyll-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine•ethyl acetate solvate.

GISADENAFIL
A mixture of N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide (1.18 kg, 2.2 Mol), potassium tert-butoxide (500 g, 4.4 moles) and ethyl acetate (193 g) in ethanol (11.8 L) was heated at 120° C. for 20 hours. The reaction mixture was then concentrated under reduced pressure, in total approx. 10 L of solvent were distilled. To the residue water (2.9 L) was added and the mixture stirred at room temperature while aqueous HCl was added until pH 7.5 was obtained. Ethyl acetate (7.5 L) was added and the two phase mixture was warmed to 55° C. The organic phase was separated and the aqueous phase was extracted with further ethyl acetate (3.0 L). The combined organic phases were distilled at atmospheric pressure to a final volume of 4 L. The precipitated solids were granulated at 5° C. for 1 h, filtered and washed with ethyl acetate (1.2 L) and dried under vacuum. This afforded 1-(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine as a light yellow crystalline solid, 877 g, 78%. m.p.=157° C. Found: C, 52.65; H, 6.46; N, 17.76. C23H33N705S. 0.2 C2H5CO2CH3 requires C, 53.21; H, 6.49; N, 18.25%.
δ (CDCl3): 1.07 (3H, t), 1.42 (3H, t), 1.61 (3H, t), 2.44 (2H, q), 2.57 (4H, m), 3.08 (2H, q), 3.15 (4H, m), 3.32 (3H, s), 3.92 (2H, q), 4.48 (2H, q), 4.77 (2H, q), 8.65 (1H, d), 9.06 (1H, d). The spectrum also has signals that correspond to a solvate with ethyl acetate.
LRMS: m/z=520 (M+1)+
.................
Example 102
1-(6-Ethoxy-5-f3-ethyll-6,7-dihvdro-2-(2-methoxyethvn-7-oxo-2r7-pyrazoler4.3- cf1pyrimidin-5-vn-3-pyridylsulfonyl)-4-ethylpiperazine»ethyl acetate solvate.

To prepare the compound of Example 8 a mixture of Λ/-[3-carbamoyl-5-ethyl- 1 -(2-methoxyethyl)-1 /-/-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1 -piperazinyl sulfonyl) nicotinamide (1.18 kg, 2.2 Mol), potassium tert-butoxide (500 g, 4.4 moles) and ethyl acetate (193 g) in ethanol (11.8 L) was heated at 120°C for 20 hours. The reaction mixture was then concentrated under reduced pressure, in total approx. 10 L of solvent were distilled. To the residue water (2.9 L) was added and the mixture stirred at room temperature while aqueous HCl was added until pH 7.5 was obtained. Ethyl acetate (7.5 L) was added and the two phase mixture was warmed to 55°C. The organic phase was separated and the aqueous phase was extracted with further ethyl acetate (3.0 L). The combined organic phases were distilled at atmospheric pressure to a final volume of 4L. The precipitated solids were granulated at 5°C for 1 h, filtered and washed with ethyl acetate (1.2 L) and dried under vacuum. This afforded 1 -(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo- 2H-pyrazole[4,3-o pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine as a light yellow crystalline solid, 877 g, 78%. m.p. = 157°C. Found: C, 52.65; H, 6.46; N, 17.76. C23H33N705S. 0.2 C2H5C02CH3 requires C, 53.21 ; H, 6.49; N, 18.25%.
δ(CDCI3): 1.07 (3H, t), 1.42 (3H, t), 1.61 (3H, t), 2.44 (2H, q), 2.57 (4H, m), 3.08 (2H, q), 3.15 (4H, m), 3.32 (3H, s), 3.92 (2H, q), 4.48 (2H, q), 4.77 (2H, q), 8.65 (1 H, d), 9.06 (1 H, d). The spectrum also has signals that correspond to a solvate with ethyl acetate.
LRMS: m/z = 520 (M+1)+
Example 103
1-(6-ethoxy-5-r3-ethyl-6.7-dihvdro-2-(2-methoxyethvn-7-oxo-2H-pyrazolor4.3- dlpyrimidin-5-vn-3-pyridylsulfonyl)-4-ethylpiperazine

GISADENAFIL
10g (0.019 mol) of the compound of Example 8 and Example 102, 1-{6- ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3- d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine ethyl acetate solvate, was charged followed by 12ml/g (120mls) of 16% water in ethyl alcohol. The slurry was heated to reflux to yield a solution and 6ml/g (60mls) distilled off at atmospheric pressure. The solution was then cooled to room temperature with crystallisation occurring at 40°C. The slurry was then cooled to 5-10°C and granulated for 30 minutes following which it was filtered and washed with 2ml/g ethyl alcohol (20 mis). The damp solid was dried in vacuo overnight at 55-60 °C to yield a white crystalline solid. (Yield 7.6g, 76%). Melting Point 162- 165°C.
δ (CDCI3): 1.05 (3H,t), 1.42 (3H,t), 1.58 (3H,t), 2.43 (2H,q), 2.57 (4H,t), 3.09 (2H, t), 3.15 (4H,t), 3.30 (3H,s), 3.93 (2H,t), 4.48 (2H,t), 4.90 (2H,q), 8.65 (1 H,d), 9.05 (1 H,d), 10.65 (1 H,s).
In the process of Example 103, water and pharmaceutically acceptable alcohols such as methanol, ethanol, propanol, butanol and mixtures thereof can be used to prepare the compound of Examples 8 and 102.
BESYLATE SALT
Example 104 1-(6-ethoxy-5-r3-ethyl-6,7-dihvdro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolor4.3- d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine benzene-sulfonate salt.

170g (0.33 mol) of the compound of Example 103, 1-{6-ethoxy-5-[3-ethyl-6,7- dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3- d]pyrimidin-5-yl]-3- pyridylsulfonyl}-4-ethylpiperazine, was charged followed by a water/ 2- butanone (4% v/v) at 10 ml/g (1.7 litres) and warmed to reflux. 53g (0.33 mol) of benzene sulfonic acid dissolved in water (23mls, resulting in 70 % w/w solution) was added to the refluxing solution over 30 minutes.5.3ml/g (0.9 litres) of 2-butanone were striped and replaced and the slurry cooled. The slurry was cooled to 5-10°C and granulated for 2 hours after which it was filtered and washed with 2ml/g (0.3 litres) of 2-butanone. The salt was dried overnight in vacuo at 55-60°C to yield a white crystalline solid. Yield 215g, 96.4%. Mpt 242-244°C. δ (DMSO): 1.17 (3H, t), 1.28 (3H, t), 1.35 (3H, t), 2.73 (2H, q), 2.97 (2H, q), 3.2 (3H, s), 3.58 (2H, t), 3.78 (3H, t), 3.81 (2H, t), 4.49 (2H, t) 4.51 (2H, q), 7.29-7.33 (3H, m), 7.57-7.60 (2H, m), 8.28 (1 H, d), 8.73 (1 H, d), 9.13 (1 H,s), 11.90(1 H,s).
The powder X-ray diffraction (PXRD) pattern for this salt, having Mpt 242- 244°C, was determined using a Siemens D5000 powder X-ray diffractometer fitted with a theta-theta goniometer, automatic beam divergence slits, a secondary monochromator and a scintillation counter. The specimen was rotated whilst being irradiated with copper K-alpha1 X-rays (Wavelength = 1.5046 Angstroms) filtered with a graphite monochromator (λ = 0.15405nm) with the X-ray tube operated at 40 kV/mA. The main peaks (in degrees θ) of the PXRD pattern are illustrated in Table I.
Table


The same besylate salt, as defined by the XRD pattern described in Table 1 , when made via alternative routes can have a melting point in the range of from 235-246°C (measured using a Perkin Elmer DSC7 at a heating rate of 20°C/minute).

References
1 The discovery of UK-369003, a novel PDE5 inhibitor with the potential for oral bioavailability and dose-proportional pharmacokinetics
Bioorg Med Chem 2012, 20(1): 498.............MP 161 - 162 °C
Bioorg Med Chem 2012, 20(1): 498.............MP 161 - 162 °C
2. Hajikarimian, Y.; Yeo, S.; Ryan, R.W.; Levett, P.; Stoneley, C.; Singh, P.
Investigation into the formation of the genotoxic impurity ethyl besylate in the final step manufacturing process of UK-369,003-26, a novel PDE5 inhibitor
Org Process Res Dev 2010, 14(4): 1027
Investigation into the formation of the genotoxic impurity ethyl besylate in the final step manufacturing process of UK-369,003-26, a novel PDE5 inhibitor
Org Process Res Dev 2010, 14(4): 1027
3. Bentham; Dawson; Dunn; Papadopoulos; Taylor; Mitchell; Snowden; Taylor
Organic Process Research and Development, 2004 , vol. 8, 4 PG. 674 - 679 .............AS ENTRY B
Organic Process Research and Development, 2004 , vol. 8, 4 PG. 674 - 679 .............AS ENTRY B
- Bloch, W., et al.: Prostate, 33, 1 (1997)
- • Glowienke, S., et al.: Mutat. Res., 581, 23 (1997)
- • Chapple, C., et al.: Eur. Urol., 54, 563 (1997)
- • Elder, D., et al.: J. Pharm. Pharmacol., 61, 269 (1997)
PATENTS
1. WO 2010062366
2. WO 2007072156
3 WO 2007072156
4.US2002/22732 A1,
5.US2002/28799 A1,
6.
| WO1998049166A1 * | Apr 10, 1998 | Nov 5, 1998 | Mark Edward Bunnage | PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3',5'-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION |
| WO1999054333A1 * | Mar 25, 1999 | Oct 28, 1999 | Mark Edward Bunnage | Pyrazolopyrimidinone cgmp pde5 inhibitors for the treatment of sexual dysfunction |
| US4666921 * | 15 окт 1985 | 19 май 1987 | Ludwig Heumann & Co. Gmbh | Pyrazole derivatives, processes for their preparation and pharmaceutical preparations containing these compounds |
| US5808092 * | 15 окт 1997 | 15 сен 1998 | Ube Industries, Ltd. | Process for preparing-1-ethyl-5-hydroxypyrazole |
| US6015911 * | 24 мар 1998 | 18 янв 2000 | Dow Agrosciences Llc | Process for preparing 1-alkyl-4-(2-chloro-3-alkoxy-4-alkylsulfonylbenzoyl)-5-hydroxypyrazole and related compounds |
| EP0463756A1 | 7 июн 1991 | 2 янв 1992 | Pfizer Limited | Pyrazolopyrimidinone antianginal agents |
| EP0812845A1 | 4 июн 1997 | 17 дек 1997 | Pfizer Limited | Process for preparing sildenafil |
| EP0994115A2 | 11 окт 1999 | 19 апр 2000 | Pfizer Limited | Process for preparation of pyrazolo-(4,3-d)pyrimidin-7-ones and intermediates thereof |
| EP0995750A1 | 15 окт 1999 | 26 апр 2000 | Pfizer Inc. | Pyrazolopyrimidinone cGMP PDE5 inhibitors for the treatment of sexual dysfunction |
| WO1998049166A1 | 10 апр 1998 | 5 ноя 1998 | Mark Edward Bunnage | PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3',5'-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION |
| WO1999054333A1 | 25 мар 1999 | 28 окт 1999 | Mark Edward Bunnage | Pyrazolopyrimidinone cgmp pde5 inhibitors for the treatment of sexual dysfunction |
| WO2001027112A1 | 4 окт 2000 | 19 апр 2001 | Charlotte Moira Norfo Allerton | 5-(2-substituted-5-heterocyclylsulphonylpyrid-3-yl)-dihydropyrazolo[4,3-d]pyrimidin-7-ones as phosphodiesterase inhibitors |
| WO2001027113A2 | 11 окт 2000 | 19 апр 2001 | Mark Edward Bunnage | PYRAZOLO `4,3-d! PYRIMIDINE DERIVATIVES |
PDE5 inhibitors mirodenafil

sildenafil

tadalafil

udenafil 3-(l-methyl-7-oxo-3-propyl-4H-pyrazolo[5,4-e]pyrimidin-5-yl)-N- [2-(l -methylpyrrolidin-2-yl)ethyl] -4-propoxybenzenesulfonamide

vardenafil 4-[2-ethoxy-5-(4-ethylpiperazin-l-yl)sulfonyl-phenyl]-9-methyl-7- propyl- 3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one

avanafil 4-[(3-chloro-4-methoxy-phenyl)methylamino]-2-[(2S)-2- (hydroxymethyl)pyrrolidin- 1 -yl] -N-(pyrimidin-2- ylmethyl)pyrimidine-5-carboxamide

dasantafil 7-[(3-bromo-4-methoxyphenyl)methyl]-l-ethyl-8-[[(lR,2R)-2- hydroxycyclopentyl]amino]-3-(2-hydroxyethyl)purine-2,6-dione

NM 702 (Nissan Chemical Industries)

SLX 101 (Surface Logix) - Structure Not Available
UK 369003 (Pfizer) - Gisadenafil besylate


No comments:
Post a Comment