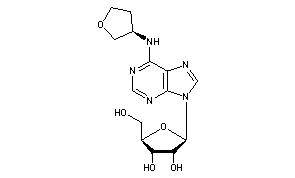
Tecadenoson
CAS : 204512-90-3
N-[(3R)-Tetrahydro-3-furanyl]adenosine
(2R,3S,4R,5R)-2-(hydroxymethyl)-5-[6-[[(3R)-oxolan-3-yl]amino]purin-9-yl]oxolane-3,4-diol
Manufacturers’ Codes: CVT-510
UNII-GZ1X96601Z; AC1L4KMO;
Molecular Formula: C14H19N5O5
Molecular Weight: 337.33
Percent Composition: C 49.85%, H 5.68%, N 20.76%, O 23.71%
Therap-Cat: Antiarrhythmic.
clinical……..http://clinicaltrials.gov/search/intervention=Tecadenoson…phase 2…………gilead
Tecadenoson is a novel selective A1 adenosine receptor agonist that is currently being evaluated for the conversion of paroxysmal supraventricular tachycardia (PSVT) to sinus rhythm. It is being developed by CV Therapeutics, Inc.
Tecadenoson is an adenosine A1 agonist which had been in phase II clinical evaluation by Gilead Sciences for treatment of atrial fibrillation. The company was also conducting phase III clinical trials for the treatment of paroxysmal supraventricular tachycardia (PSVT); however, no recent developments have been reported for these indications.
Due to the fact that tecadenoson selectively stimulates the A1 receptor and slows electrical impulses in the heart’s conduction system without significantly stimulating the A2 receptor, the intravenous administration of CVT-510 may hold potential for rapid intervention in the control of atrial arrhythmias without lowering blood pressure.


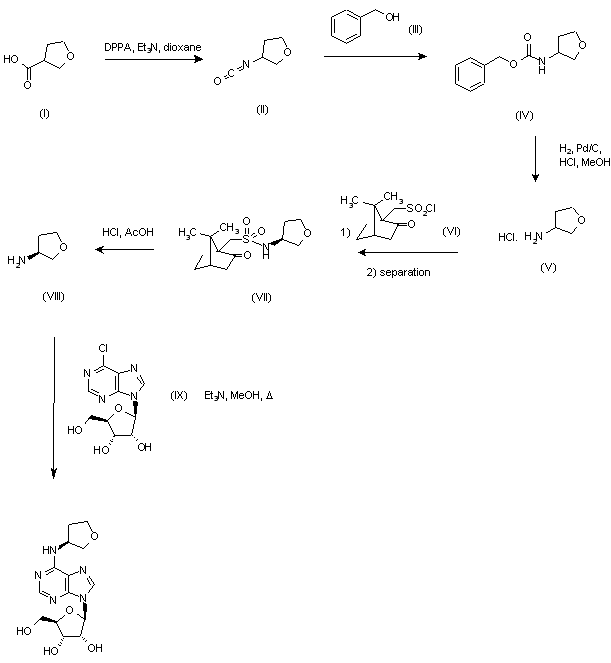
The reaction of 3-tetrahydrofuroic acid (I) with diphenyl phosphoryl azide (DPPA) in refluxing dioxane gave the intermediate isocyanate (II), which was treated with benzyl alcohol (III) to yield carbamate (IV). Subsequent hydrogenolysis in the presence of Pd/C afforded racemic amine (V), which was resolved by treatment with S-(+)-10-camphorsulfonyl chloride (VI) in pyridine, followed by column chromatography and recrystallization from acetone of the resulting sulfonamide (VII). Then, hydrolysis in HCl-AcOH provided the S-amine (VIII). Condensation of amine (VIII) with 6-chloropurine riboside (IX) in the presence of triethylamine in refluxing MeOH furnished the title compound.
EP 0920438; EP 0992510; JP 2000501426; US 5789416; WO 9808855
……………………………
………………………….
CVT-510 (tecadenoson) has chemical structure (8 :

…………………………………….
Compound I can be prepared through reaction of the corresponding primary amino compound, R1NH2, through heating with commercially available 6-chloroadenosine in the appropriate solvent (e.g. n-butanol, dimethylformamide, and ethanol). The primary amino compound, R1NH2, is either commercially available or can be prepared as previously described (International Patent Application WO 98/08855).

……………………………
EXAMPLE 1
The compounds of this invention may be prepared by conventional methods of organic chemistry. The reaction sequence outlined below, is a general method, useful for the preparation of compounds of this invention.


According to this method, oxacycloalkyl carboxylic acid is heated in a mixture of dioxane, diphenylphosphoryazide and triethylamine for 1 hour. To this mixture is added benzyl alcohol and the reaction is further heated over night to give intermediate compound 1. Compound 1 is dissolved in methanol. Next, concentrated HC1, Pd/C is added and the mixture is placed under hydrogen at 1 atm. The mixture is stirred overnight at room temperature and filtered. The residue is recrystallized to give intermediate compound 2. 6-chloropurine riboside is combined and the mixture is compound 2 dissolved in methanol and treated with triethylamine. The reaction is heated to 80° C for 30 hours. Isolation and purification leads to Compound 3.
EXAMPLE 2
Compounds of this invention prepared according to the method of Example 1 were tested in two functional models specific for adenosine A, receptor agonist function. The first was the A , receptor mediated inhibition of isoproterenol stimulated cAMP accumulation in DDT cells. The EC50 of each derivative is shown in Table I. Also shown in Table I is the ability of each derivative to stimulate cAMP production in PC 12 cells, a function of agonist stimulation of adenosine A2 receptors. The ratio of the relative potency of each compound in stimulating either an A, receptor or an A2 receptor effect is termed the selectivity of each compound for the A, receptor. As can be seen in Table I, each derivative is relatively selective as an A, receptor agonist. The use of measuring cAMP metabolism as an assay for adenosine A , receptor function has been previously described (Scammells, P., Baker, S., Belardinelli, L., and Olsson, R. , 1994, Substituted 1 ,3-dipropylxanthines as irreversible antagonists of A, adenosine receptors. J. Med. Chem 37: 2794-2712, 1994).
Table I
Compound R EC50 (nM) ECS, (nM) A,/A2 A-/A, DDT cells PC 12 cells
I 4-arninopyran 12 970 0.012 80.0
II (±)-3-aminotetrahydrofuran 13 1400 0.0093 107.6
III (R)-3-aminotetrahydrofuran 1.08 448 0.0024 414
IV ( 1 )-caprolactam 161 181 0.889 1.12
V (S)-3-aminotetrahydrofuran 3.40 7680 0.00044 2258
Compounds were also tested in a whole organ model of A, receptor activation with respect to atrial and AV nodal function. In this model, guinea pig hearts are isolated and perfused with saline containing compound while atrial rate and AV nodal conduction time are assessed by electrographic measurement of atrial cycle length and AV intervals, as detailed in Belardinelli, L, Lu, J. Dennis, D. Martens, J, and Shryock J. (1994); The cardiac effects of a novel A,-adenosine receptor agonist in guinea pig isolated heart. J. Pharm. Exp. Therap. 271:1371-1382 (1994). As shown in Figure 1, each derivative was effective in slowing the atrial rate and prolonging the AV nodal conduction time of spontaneously beating hearts in a concentration-dependent manner, demonstrating efficacy as adenosine A, receptor agonists in the intact heart.
EXAMPLE 3
Preparation ofN-benzyloxycarbonyl-4-aminopyran.
A mixture of 4-pyranylcarboxylic acid (2.28 gm, 20 mmol), diphenylphosphorylazide (4.31 ml, 20 mmol), triethylamine (2.78 ml, 20 mmol) in dioxane (40 ml) was heated in a 100° C oil bath under dry nitrogen for 1 hour. Benzyl alcohol (2.7 ml, 26 mmol) was added, and heating was continued at 100° C for 22 hours. The mixture was cooled, filtered from a white precipitate and concentrated. The residue was dissolved in 2N HC1 and extracted twice with EtOAc. The extracts were washed with water, sodium bicarbonate, brine and then dried over MgSO4, and concentrated to an oil which solidified upon standing. The oil was chromatographed (30% to 60% EtO Ac/Hex) to give 1.85 g of a white solid (40%).
Preparation of 4-aminopyran.
N-benzyloxycarbonyl-4-aminopyran (1.85 gm, 7.87 mmol) was dissolved in MeOH (50 ml) along with cone. HC1 and Pd-C ( 10%, 300 mg). The vessel was charged with hydrogen at 1 atm and the mixture was allowed to stir for 18 hours at room temperature. The mixture was filtered through a pad of eelite and concentrated. The residue was co-evaporated twice with MeOH/EtOAc and recrystallized from MeOH/EtOAc to afford 980 mg (91 %) of white needles (mp 228-230° C).
Preparation of 6-(4-aminopyran)-purine riboside. A mixture of 6-chloropurine riboside (0.318 gm, 1. 1 mmol), 4-aminopyran-HCl
(0.220 mg,
1.6 mmol) and triethylamine (0.385 ml, 2.5 mmol) in methanol (10 ml) was heated to 80° C for 30 hours. The mixture was cooled, concentrated and the residue chromatographed (90: 10: 1, CH2 Cl2/MeOH/PrNH2). The appropriate fractions were collected and recliromatographed using a chromatotron
(2 mm plate, 90: 10: 1, CH2 Cl2/MeOH/PrNH2) to give an off white foam (0.37 gm, 95%).
EXAMPLE 4
Preparation of N-benzyloxycarbonyl-3-aminotetrahydrofuran. A mixture of 3-tetrahydrofuroic acid (3.5 gm, 30 mmol), diphenylphosphorylazide (6.82 ml, 32 mmol), triethylamine (5 ml, 36 mmol) in dioxane (35 ml) was stirred at RT for 20 min then heated in a 100° C oil bath under dry nitrogen for 2 hours. Benzyl alcohol (4.7 ml, 45 mmol) was added, and continued heating at 100° C for 22 hours. The mixture was cooled, filtered from a white precipitate and concentrated. The residue was dissolved in 2N HC1 and extracted twice using EtOAc. The extracts were washed with water, sodium bicarbonate, brine dried over MgSO4, and then concentrated to an oil which solidifies upon standing. The oil was chromatographed (30% to 60% EtO Ac/Hex) to give 3.4 g of an oil (51
%).
Preparation of 3-aminotetrahydrofuran.
N-benzyloxycarbonyl-3-aminotetrahydrofuran (3.4 gm, 15 mmol) was dissolved in MeOH (50 ml) along with cone. HC1 and Pd-C (10%, 300 mg). The vessel was charged with hydrogen at 1 atm and the mixture was allowed to stir for 18 hours at room temperature. The mixture was filtered through a pad of celite and concentrated. The residue was co-evaporated two times with MeOH/EtOAc and recrystallized from MeOH/EtOAc to give 1.9 g of a yellow solid.
Preparation of 6-(3-aminotetrahydrofuranyl)purine riboside. A mixture of 6-chloropurine riboside (0.5 gm, 1.74 mmol), 3-aminotetrahydrofuran
(0.325 gm, 2.6 mmol) and triethylamine (0.73 ml, 5.22 mmol) in methanol (10 ml) was heated to 80° C for 40 hours. The mixture was cooled, and concentrated. The residue was filtered through a short column of silica gel eluting with 90/10/1 (CH2Cl2/MeOH/PrNH2), the fractions containing the product were combined and concentrated. The residue was chromatorgraphed on the chromatotron (2 mm plate, 92.5/7.5/1 , CH2CL2/MeOH/P.NH2). The resulting white solid was recrystallized from MeOH/EtOAc to give 0.27 gm of white crystals (mp 128-130° C).
EXAMPLE 5
Resolution of 3-arninotetrahydrofuran hydrochloride
A mixture of 3-aminotetrahydrofuran hydrochloride (0.5 gm, 4 mmol) and
(S)-(+)-10-camphorsulfonyl chloride (1.1 gm, 4.4 mmol) in pyridine (10 ml) was stirred for 4 hours at room temperature and then concentrated. The residue was dissolved in EtOAc and washed with 0.5N HC1, sodium bicarbonate and brine. The organic layer was dried over MgSO4, filtered and concentrated to give 1. 17 g of a brown oil (97%) which was chromatographed on silica gel (25% to 70% EtOAc/Hex). The white solid obtained was repeatedly recrystallized from acetone and the crystals and supernatant pooled until an enhancement of greater than 90% by 1H NMR was acheived.
Preparation of 3-(S)-aminotetrahydrofuran hydrochloride.
The sulfonamide (170 mg, 0.56 mmol) was dissolved in cone. HCl/AcOH (2 mL each), stirred for 20 hours at room temperature, washed three times with CH2C12 (10 ml) and concentrated to dryness to give 75 mg (qaunt ) of a white solid
Preparation of 6-(3-(S)-aminotetrahydrofuranyl)puπne riboside.
A mixture of 6-chloropurιne riboside (30 mg, 0.10 mmol),
3-(S)-amιnotetrahydrofuran hydrochloride (19 mg, 0.15 mmol) and triethylamine (45 ml, 0.32 mmol) in methanol
(0.5 ml) was heated to 80° C for 18 hours. The mixture was cooled, concentrated and chromatographed with 95/5 (CH2Cl /MeOH) to give 8 mg (24%) of a white solid.
Literature References:
Selective adenosine A1-receptor agonist. Prepn: R. T. Lum et al., WO 9808855; eidem, US5789416 (both 1998 to CV Therapeutics).
Clinical effect on AV nodal conduction: B. B. Lerman et al., J. Cardiovasc. Pharmacol. Ther.6, 237 (2001).
Clinical evaluation in paroxysmal supraventricular tachycardia: E. N. Prystowsky et al., J. Am. Coll. Cardiol. 42, 1098 (2003); K. A. Ellenbogen et al., Circulation 111, 3202 (2005).
Review of pharmacology and clinical experience: A. Zaza, Curr. Opin. Invest. Drugs 3, 96-100 (2002); J. W. Cheung, B. B. Lerman, Cardiovasc. Drug Rev. 21, 277-292 (2003).
| US7144871* | 19 Feb 2003 | 5 Dec 2006 | Cv Therapeutics, Inc. | Partial and full agonists of A1 adenosine receptors |
| US7696181* | 24 Aug 2006 | 13 Apr 2010 | Cv Therapeutics, Inc. | Partial and full agonists of A1 adenosine receptors |
Keywords: Antiarrhythmic, Adenosine Receptor Agonist, Tecadenoson, CVT-510, CV Therapeutics
No comments:
Post a Comment